آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Nucleic acid Amplification Techniques |


|
|
Read More
Date: 14-12-2015
Date: 11-12-2020
Date: 8-5-2016
|
Nucleic acid Amplification Techniques
1- Polymerase Chain Reaction (PCR)
Excellent reviews on its applicability in clinical microbiology have been published.Due to its extreme sensitivity, nucleic acid amplification has revolutionised the identification of infectious agents in patient samples. Several alternative nucleic acid amplification methods have been developed for use in the clinical microbiological laboratory, such as the ligase chain reaction (LCR), isothermal strand displacement amplification (SDA) and isothermal transcription-mediated amplification (TMA/NASBA).
2- The Contamination Problem
Great care must be taken in any diagnostic laboratory to minimise the problem of contamination of samples due to the exquisite sensitivity of nucleic acid amplification techniques. Sterile filter tips must be employed for all manual pipetting steps. The logistics of the laboratory procedures are of fundamental importance. A separate room should be reserved for work with buffer components, oligonucleotide primers and probes and mastermixes. No samples or amplified products should be allowed into this room. Higher air pressure than in the surrounding rooms, restricted access, visible written hygiene guidelines on the door and use of separate clothing and shoes are advantageous. A separate room is necessary for receiving and organising patient samples and a third room for setting up the PCR reactions. Using conventional PCR, a fourth separate room will be necessary for the detection steps of the amplified products, the amplicons. Commonly used methods for the visualisation of amplicons
are agarose gel electrophoresis and microplate systems with a detection probe and a streptavidin–enzyme conjugate that binds to biotinylated nucleotides incorporated into the amplicon. Following a washing step, the enzyme bound by the biotin–streptavidin interaction will change a colourless substrate into a coloured derivative in a quantitative way. Using real-time PCR, the amplification and detection can be completed in a closed system and for space considerations may be performed in the same room as the reaction setup. If possible, one-way flow of persons and samples from the first to the last room is desirable. Decontamination of critical work space using UV light or 5% bleach represents an additional precaution against contamination of samples.When UTP is used in the deoxynucleotide mix of the amplification buffer of the previous setup, a preamplification step using uracil-N-glycosylase will efficiently remove amplicon contamination in the subsequent reaction.
3- Reverse PCR – cDNA Synthesis
DNA is suitable for diagnostic purposes when the infectious agent is a bacterium, a parasite or a DNA virus. A large proportion of viruses are RNA viruses, however, i.e. their genomes consist of RNA and are replicated in host cells as RNA. Reverse transcription of RNA to complementary DNA (cDNA) is therefore an obligatory step for the identification of RNA viruses. When PCR DNA amplification is preceded by cDNA synthesis, the combined protocol is named reverse PCR. Reverse PCR may also be useful in order to quantify transcription (mRNA synthesis) to show the activity of an infectious genome. Using enzymes such as rTth, which has both reverse transcriptase and DNA polymerase activity and is thermostable, reverse transcription can be achieved in one reaction and one tube.
Otherwise, the reverse transcription is often carried out as a separate first step using a thermolabile reverse transcriptase (typically cloned retroviral enzymes). If random primers (hexamers or nonamers) or oligo-dT primers are used in the initial reverse transcription, then the subsequent PCR can be performed with different specific primer pairs and designed for different RNA viruses. According to the rTth reverse PCR, the same reverse primer sequence will prime both cDNA synthesis and the PCR amplification. This may increase specificity, but reduces the versatility compared with random hexamer or oligo-dT primed cDNA.
4.4.4 Nested PCR
Experience shows that whenever PCR is performed on a patient sample, the amplification products very often exhibit a smear in the following ethidium bromide-stained agarose gel electrophoresis. This is due to nonspecific priming in the background of high amounts of host cell nucleic acids and the need to perform a large number of thermocycles (typically 40). Although the first PCR amplification resulted in a smear in the agarose gel, the specific nucleic acids sought will usually be highly enriched compared with the starting material. If a small amount of the first PCR is used for a second PCR amplification with a new set of internal primers, then a well-defined PCR fragment typically results if the infectious agent is present in the initial sample. The use of a second round of PCR with a new set of internal primers is called a nested PCR. If only one internal primer is used for the second round of thermocycling, it is called a semi-nested PCR.
Nested and semi-nested PCR therefore increase the sensitivity as much as 1000-fold compared with conventional PCR9 and increase specificity, but the techniques require more work and longer assay times and carry an increased risk of contamination.
5- Real-time PCR
Compared with conventional PCR, and in particular compared with nested PCR, real-time PCR has the advantage that the amplification process is recorded in real time. The risk of contamination is very much reduced since there is no need to open the reaction tube once the amplification has started, i.e. the post-amplification detection step is omitted. Therefore, and because more of the process can be automated, it takes much less time and labour to do real-time PCR than nested PCR. For the same reasons, the risk of sample switching is lower in realtime PCR. The sensitivity is comparable between real-time PCR and nested PCR. Real-time PCR results are digitised instantly and analysed using standardised computer algorithms, thus minimising human errors, and may provide test results within 1 h after the start of the analysis, whereas analysis based on nested PCR may take hours to complete.
6- Visualisation of Real-time PCR Amplification
The critical difference between conventional and real-time PCR is that in the latter the generation of amplification products is monitored directly in the reaction tube. Different fluorescence-based principles are employed for real-time detection. SYBR Green represents one class of DNA binding chemicals that emits much more fluorescence when intercalated into double-stranded DNA compared with the unbound chemical in solution (Figure 1). As a result, the SYBR Green fluorescence increases proportionally to the increase in DNA amplification products. The disadvantage of using SYBR Green is that non-specifically amplified DNA and amplification artefacts such as primer dimers will also generate increased fluorescence, thus reducing the sensitivity compared with nested PCR. Real-time PCR assays utilising an amplicon- specific probe increase the specificity substantially. A number of ‘smart’ assays have been invented based on three oligonucleotides,i.e. two primers and a probe. Several of these assays include TaqMan assays (Figure .2), fluorescence resonance energy transfer (FRET) (Figure .3) assays, Eclipse probes, Scorpion probes and molecular beacon probes.
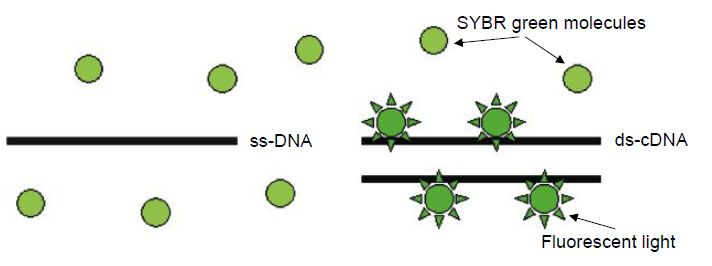
Figure 1 The SYBR Green molecule has high affinity to double-stranded DNA. SYBR Green fluorescence is much stronger when it is bound to DNA compared with unbound SYBR Green. ss-DNA=single-stranded DNA; ds-DNA=double-stranded DNA.
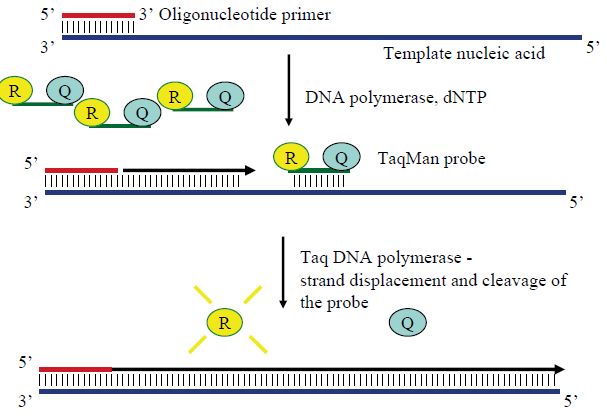
Figure 2 The principle of TaqMan real-time PCR assays. The probe is labelled with both a fluorescent chemical group (R) and a quencher (Q). When the Taq polymerase elongates the complementary strand from the primer, its 5´–3´ exonuclease activity cleaves any annealed probe, thus separating R from Q. Freed from its quencher, the R fluoresces when exciting light is sent in. The amount of free R, and thus fluorescence, increase exponentially along with amplicons until the PCR reaction approaches the plateau stage.
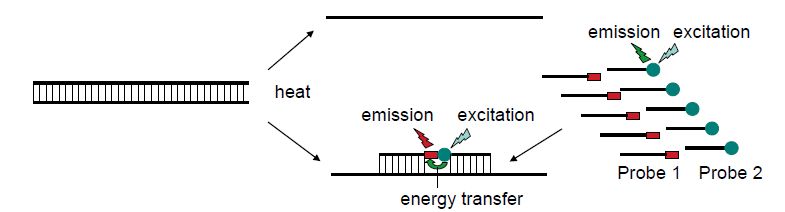
Figure 3 The principle of fluorescence resonance energy transfer (FRET). Two different probes are complementary to the target sequence. One probe is labelled with fluorescein and the other with LC Red fluorochrome. The hybridisation to the target sequence will bring the two fluorochromes physically close enough for the emission energy of fluorescein to excite the LC Red so that red fluorescent light is emitted. When PCR generates an increasing concentration of target, an increasing fraction of the probes can be positioned to achieve FRET. This can be monitored as increasing red fluorescence.
7- Real-time PCR Equipment
Compared with conventional PCR, real-time qPCR puts extra demands on the equipment. Lasers are needed to excite fluorophores and recording devices are required to collect and digitise the fluorescent signals. Several of the available thermocyclers are listed in Table 1 in Ref. 2. The choice of thermocycler will depend on a large number of variables. Should the thermocycler be downstream of a high-throughput 96-well-format? Or is there a need for individualisation of PCR reactions that can be provided by the Smartcycler?
8- Real-time Quantitative PCR
The increasing fluorescence signal plotted on the y-axis and the increasing thermocycle numbers along the x-axis generate a sigmoidal curve. The threshold cycle is defined as the cycle number when the fluorescent signal exceeds the detection threshold. During the following 4–8 cycles, the fluorescence typically approaches an exponential increase before approaching a plateau phase. During the exponential phase, the logarithm of the fluorescent signal along the y-axis and the cycle number along the x-axis generate a straight line that can be used for quantification. In this phase of the real-time PCR, the fractional threshold cycle number (Ct) is inversely related to the logarithm of the number of starting templates. Parallel PCR amplification of a dilution series of known amounts of the same template therefore allows the construction of a calibration curve from which the exact amount of starting template can be calculated. It must then be assumed that the amplification efficiency is equal for all samples and standards. One sign of acceptable quantification is that the slope of the calibration curve is between -3.3 and -3.4. More exact guidelines to quantification and quality control can be found in user bulletins to thermocycler software (e.g. ABI Prism User Bulletin No. 2, http://www.appliedbiosystems.com/).
8.1 - Absolute and Relative Quantifications
In order to achieve absolute quantification, a known amount of reference standard is required for the dilution series and calibration curve generation. Synthetic oligonucleotides, purified PCR fragments or plasmids or in vitro transcribed RNA can be quantified using absorption readings at a wavelength of 260nm (OD260). Dilution series of exactly known amounts of target then allow absolute quantification. Relative
quantification, relating the amount of template to an accepted standard, is often sufficient and this can be achieved using the calibration curve method or the comparative Ct method .
9- Determination of ‘Viral Load’ in Clinical Microbiology
‘Viral load’ refers to the number of viral genomes in a defined volume of a patient sample, typically in serum or plasma. Determination of viral load is becoming increasingly important with increasing use of antiviral medication.
Real-time quantification of viral load is one important criterion for the start of antiviral medication against human immunodeficiency virus (HIV), hepatitis C and hepatitis B viruses, against cytomegalovirus (CMV) in immunocompromised patients and against Epstein–Barr virus (EBV) in post-transplant patients. Efficient response to antiviral therapy can be monitored as the viral load is reduced and quantification is of great value to monitor possible development of viral resistance to the medication or possible relapse after cessation of therapy. Commercial kits are widely used for quantification of viral load in clinical microbiology. A known amount of synthetic standard is typically included in each reaction to establish a competitive PCR amplification. The resulting amounts of viral and standard amplicons are compared with a standard dilution curve in order to establish the viral load. It is important to be aware that different manufacturers may have different standardisation units so that results obtained using different kits may not be comparable. Progress has recently been made to establish internationally accepted standard units for different viruses, e.g. for hepatitis B virus,hepatitis C virus and parvovirus B19. The WHO provides guidelines and established standards in order to calibrate different methods to obtain comparable International Units per millilitre (IUmL-1) (http://www.who.int/biologicals/reference_
preparations/distribution/en/index.html).
10- Internal Controls in Microbiological Real-time qPCR
Both negative and positive controls are important in nucleic acid-based diagnosis in the microbiological laboratory. The negative control consists of a parallel amplification reaction that does not contain the specific target. The most difficult decision is the number of negative controls and recommendations for a negative control for every fifth tube to control efficiently for contamination may come in conflict with available resources. Positive controls can be any target sequence in a separate tube. The best positive control templates are included in the same tube as the diagnostic assay and contain flanking sequences complementary to the primers of the diagnostic assay but with a unique sequence in the region recognised by the probe. Since real-time qPCR
assays are typically designed with amplicons shorter than 100 nucleotides, it is easy to obtain the desired control template sequence by oligonucleotide synthesis. A more difficult challenge is if the laboratory wants to spike the patient sample with a control sequence to control for the entire process from sample handling, via nucleic acid extraction through PCR amplification. For RNA viruses, which represent the greatest challenge, this can be achieved if a known amount of an in vitrosynthesised RNA is spiked into the patient sample. Any exogenous sequence will do if it can be detected separately from the sequence of the infectious agent. However, the optimal control RNA will be transcribed from a vector that can generate an RNA that contains more than 1500 nucleotides and includes a target sequence that can be amplified with the
same primers as the diagnostic assay but differs in the region recognised by the probe. In all same-tube controls it is very important to titrate carefully the amount of control target. Otherwise, the control may compete out the diagnostic assay and lead to false negatives.
11- Multiplex Real-time PCR
Multiplex PCR refers to the use of more than one primer pair in one reaction tube in order to amplify more than one target sequence or to more than one primer/probe set in the case of real-time PCR. The internal control assay described above is one example of multiplex PCR. In microbiological diagnostics, the general aim of multiplex PCR is to test simultaneously for different agents in the same reaction. This requires the use of fluorophores and equipment with the ability to distinguish between the different assays. Non-specific amplification products seem to be the most limiting factor of multiplex PCR as the number of oligonucleotide primers and probes increases. Several multiplex assays for the simultaneous detection of two or more different agents are, however, well established. Examples include multiplex PCR assays for herpes simplex virus 1 and 2 (HSV-1 and HSV-2) and assays for influenza type A and B viruses.
4.4.12 Melting Curve Analysis
By taking advantage of melting curve analysis, it has been possible to increase the number of agents detected in multiplex real-time PCR, in particular to distinguish between closely related viruses such as HSV-1 and HSV-2 or influenza A and B. FRET probes (Figure 3) are useful for this type of analysis. When one FRET probe is designed to be complementary to one viral subtype but contains one or very few mismatches to the other viral subtype, melting curve analysis is possible due to different melting points (Tm) depending on the exact target sequence. Melting curve analysis is performed following completion of the PCR. The probe pair is allowed to anneal to the target amplicons and then the temperature is increased while the fluorescence is continuously recorded. The viral subtype with mismatch to the FRET probe will exhibit a reduced fluorescence at a lower Tm than the viral subtype with perfect homology to the FRET probe. If both subtypes are present in the same mix, this may be visualised as a curve with two different peaks when the fluorescence is plotted on the y-axis and temperature on the x-axis. A number of multiplex real-time PCR assays have been published, including simultaneous amplification and subtyping of HSV-1 and HSV2 and VZV using FRET probes and the combination of influenza A subtypes H1N1 and H3N2, influenza B and respiratory syncytial virus (RSV) subtypes A and B.
13- Genotyping
Viruses evolve much more rapidly than their host organisms. In particular, RNA viruses that are replicated by RNA polymerases which lack 5´ proof reading, in contrast to DNA polymerases, are prone to point mutation errors in the order of 10-4 per genome replication. Both DNA viruses and RNA viruses may change rapidly when there are changes in the selection pressure due to very short generation times and an enormous number of progeny in each infection. As a consequence, most viral species come in multiple serotypes and genotypes and we have the dubious possibility to eyewitness viral evolution. In many cases it is clinically relevant to distinguish between genotypes. Twelve different main genotypes are currently described for hepatitis C virus. Both prognosis and the recommended duration of PEG-interferon a-ribavirin combination therapy vary between HCV genotypes. The prevalent genotypes 1 and 4 usually require 48 weeks of combination therapy whereas 12 weeks of therapy is often sufficient for genotypes 2 and 3. Also, sustained viral response, meaning that HCV RNA cannot be detected in serum 6 months following cessation of treatment, is achieved in a higher proportion of cases for genotypes 2 and 3. More than 130 genotypes of human papillomaviruses (HPVs) are currently recorded. Less than 90% sequence homology in regions of the L1, E6 and E7 genes compared with any previously known HPV type is required to define a new HPV type. HPVs are separated into high- and low-risk types regarding their ability to cause cervical cancer. Genotyping can be achieved using DNA sequencing. Due to the current labour and cost of DNA sequencing, other methods have been developed for genotyping in the clinical microbiology laboratory such as sandwich hybridisation assays (Figure 4) and line probe assays (Figure 5).
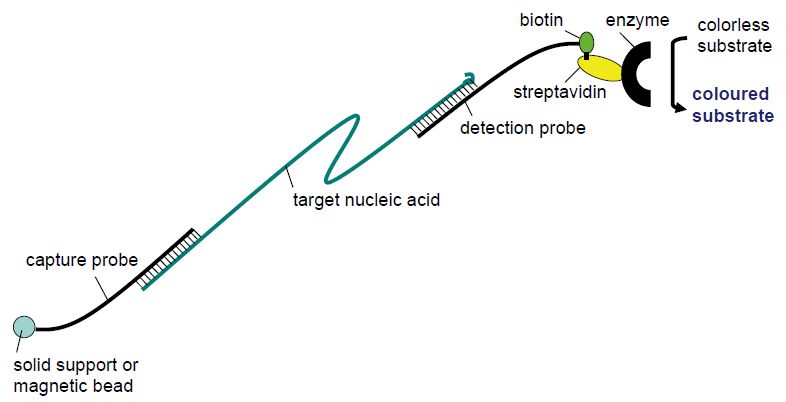
Figure 4 Principle of sandwich hybridisation.
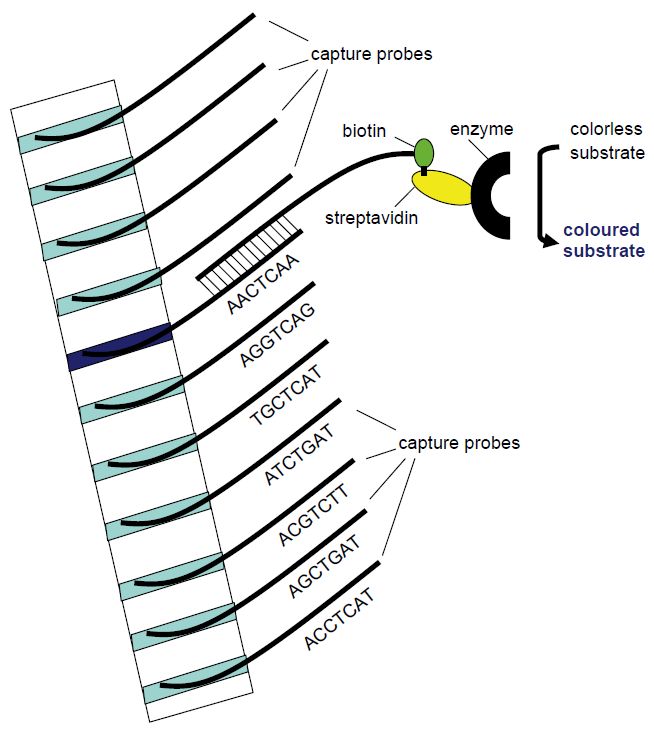
Figure 5 Principle of the line probe assay. Different capture probe sequences are
fixed in different bands in a membrane strip. The amplification product will hybridise with the capture probe of complementary sequence. Utilising biotin-labelled PCR primers, the position of the complementary capture probe can be visualised as a coloured band following addition of streptavidin-bound enzyme that will change a non-coloured substrate to a coloured product that precipitates in the band.

|
|
|
|
إجراء أول اختبار لدواء "ثوري" يتصدى لعدة أنواع من السرطان
|
|
|
|
|
|
|
دراسة تكشف "سببا غريبا" يعيق نمو الطيور
|
|
|
|
|
|
لأعضاء مدوّنة الكفيل السيد الصافي يؤكّد على تفعيل القصة والرواية المجسّدة للمبادئ الإسلامية والموجدة لحلول المشاكل المجتمعية
|
|
|
|
قسم الشؤون الفكرية يناقش سبل تعزيز التعاون المشترك مع المؤسّسات الأكاديمية في نيجيريا
|
|
|
|
ضمن برنامج عُرفاء المنصّة قسم التطوير يقيم ورشة في (فنّ الٕالقاء) لمنتسبي العتبة العباسية
|
|
|
|
وفد نيجيري يُشيد بمشروع المجمع العلمي لحفظ القرآن الكريم
|