آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Bioinformatics |


|
|
Read More
Date: 12-5-2016
Date: 1-1-2016
Date: 21-5-2021
|
Bioinformatics
Bioinformatics is a new branch of molecular biology, also kown as computational biology. Computational physics and computational chemistry have emerged as third branches in their respective disciplines, after the experimental and theoretical branches, due largely to the advance of computational capabilities in modern computers. In contrast, bioinformatics has emerged in molecular biology as the result of advances in experimental technology, especially in the high-throughput DNA sequencing, which is generating a vast amount of gene and protein sequence data. Since initiation of the Human Genome Project in the late 1980s, bioinformatics has become an integral part of the coordinated efforts to sequence the entire genomes of a number of organisms from bacteria to human. Consequently, bioinformatics is a data-driven discipline requiring large-scale databases and associated technologies for data management and interpretation. This contrasts with the model-driven, theory-based approaches in computational physics and computational chemistry.
Bioinformatics covers a diverse range of topics, which broadly have two roles. First, bioinformatics is widely used in experimental projects, such as in determining the optimal genetic mapping strategy and how best to assemble raw sequence data, and in the storing and handling information and materials. Secondly and more important, the major task of bioinformatics is to develop new databases and new computational technologies that will help to understand the biological meaning encoded in the sequence data. Interpretation of sequence data cannot be achieved by numerical calculations based on first-principle equations, which are virtually nonexistent in biology, but it requires using and processing empirical knowledge acquired from experimental data. This is what human experts would do if the amount of data and knowledge were manageable in size. Thus, bioinformatics was born from the marriage of molecular biology, in the age of massive data production, with artificial intelligence, which is a mature branch of computer science, to automate knowledge processing.
Table 1 summarizes topics in bioinformatics for interpretating of sequence data. The methods of computer science have been used traditionally in artificial intelligence applications, such as speech recognition and natural language processing, but they are also quite effective in solving problems in molecular biology. There is a rough distinction between how data are organized and consequently what kinds of computational methods are used in the three categories of Table 1. In the first category of similarity searches, the database is a collection of all the known primary data, such as the sequence database and the structure database, which are the repositories of all reported sequences and three-dimensional structures, respectively, of nucleic acids and proteins. A basic operation in this category is comparing of individual sequences or structures to detect any similarity. For example, to understand the functional implications of a newly determined gene or protein sequence, it is customary to perform a sequence similarity, or homology, search, comparing the query sequence with each of the sequences in the database. If any similar sequence is found in the database, the query sequence is assumed to have a similar or related function. This reasoning is based on the empirical observation that homologous sequences generally share similar functions and similar 3-D structures, because they arose from a common evolutionary ancestor and because of functional constraints.
Table 1. Bioinformatics for Interpretation of Sequence Data

The procedure of comparing two sequences is a problem of sequence alignment, which is a major topic in sequence analysis. Given a similarity measure of amino acids or nucleotides, the problem of obtaining the best sequence alignment is equivalent to optimizing a given score function that represents the overall similarity. There are variations in how the optimal alignment is made, either globally for the entire sequences or locally to detect localized regions, how many sequences are aligned at a time, and which similarity measure is to be used. By allowing insertions and deletions (indels), the number of possible alignments grows exponentially as the number and length of the sequences to be compared increases. Thus, the sequence alignment problem is a typical combinatorial optimization problem in computer science. Although the alignment of two or three sequences can be solved rigorously by using dynamic programming algorithms (1), which effectively evaluate all possibilities, multiple alignment of many sequences requires heuristic algorithms to obtain approximate solutions. Furthermore, to search large databases effectively for similar sequences, heuristic algorithms such as FASTA (2) and BLAST (3) have been developed for rapidly identifying regions of two sequences that are similar locally. Other extensions of the sequence alignment algorithms include analysis of a single RNA sequence to predict any secondary structure and compare two 3-D protein structures.
When humans try to interpret a sentence written in a foreign language, they use dictionaries and a knowledge of grammar. However, the process of using a homology search to interpret a sentence written in the DNA language is like comparing it with all the sentences written in the past and checking precedents as to how they were interpreted. Sequence interpretation would become more efficient if our knowledge of sequence-functional relationships were more advanced. The second and third categories in Table 1 involve such reorganizations of the primary data. In the second category of structure/function prediction, the primary data are classified into a group based on a given criterion, such as functional identity, and higher level knowledge is abstracted from this group. This is based on our empirical observation that the function of a protein molecule is usually exerted at a functional site in its three-dimensional structure, which is usually a ligand-binding site for other molecules, and that the site is formed by one or more linear polypeptide segments that have conserved sequence patterns. These conserved patterns are called sequence motifs, and they can be extracted, for example, by multiple alignment of a set of sequences that have the same function. A motif search against a library of known motifs is widely used as an alternative method for interpreting sequence data, when no similar sequences have been found by a homology search. However, the bioinformatics problem here is not how to search the known motifs, but how best to extract and define new motifs effectively. Thus, the problem is related to machine learning in computer science, where a higher level concept or a generalized pattern is abstracted from a set of given examples.
Although the second category in Table 1 is targeted at a specific function and a specific group of sequences, the third category of molecular classification represents a more global analysis, where the grouping is attempted with all the available data, and the relationship among groups, which is often hierarchical, is examined. For example, all known protein sequences can be hierarchically classified into families and superfamilies according to their level of sequence similarity. Because complete genome sequences are available, it has become possible to establish similarity relationships of all genes within species (paralogous genes) and across species (orthologous genes). These analyses are carried out by clustering algorithms, and the similarity score is used as a measure of the closeness of two sequences. In practice, however, there are biological difficulties, notably due to the abundance of multidomain, multifunctional proteins, which make automatic clustering a complicated task.
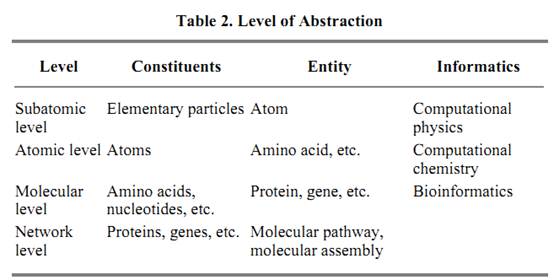
Molecular biology has been dominated by a reductionist approach where, starting from a selected functional aspect of life, such as metabolism, signal transduction, or development, molecular components and their interactions are identified experimentally. In contrast, sequencing an entire genome identifies a complete set of genes and proteins in an organism, but it does not tell how they should be interrelated to form a functioning, living system. This is where a synthetic approach based on bioinformatics will play an increasingly more dominant role in the basic research into understanding life and in the applied research into biomedical relevance. Table 2 summarizes the role of bioinformatics relative to the level of abstraction in nature. At the molecular level, an amino acid residue is represented by a symbol, for example, C for cysteine, which is a compound consisting of carbon, hydrogen, nitrogen, oxygen, and sulfur atoms. When comparing two amino acid sequences, only the one-dimensional connection of symbols is considered biologically relevant, neglecting any atomic details. At a higher level of abstraction, which may be called a network level, a gene or a protein is represented by a symbol, for example, Ras for an oncogene product. A primary concern here is the Ras signal transduction pathway, which is generated by a network of interacting molecules. Bioinformatics is relatively well established at the molecular level, such as in the sequence and structure databases and in the computational methods to analyze them. To step up to a
higher level, however, it is necessary to develop new databases and new computational technologies, such as a network database containing all the wiring-diagram information about genes and molecules, plus the algorithms to analyze them. This direction would eventually lead to in silico reconstruction of a biological organism.
References
1.S. B. Needleman and C. D. Wunsch (1970) J. Mol. Biol. 48, 443–453.
2.W. J. Wilbur and D. J. Lipman (1983) Proc. Natl. Acad. Sci. USA 80, 726–730.
3. S. F. Altschul et al. (1990) J. Mol. Biol. 215, 403–410.

|
|
|
|
صنع الذكريات والتفكير يدمر الدماغ.. دراسة تشرح السبب
|
|
|
|
|
|
|
بركان ينفت الذهب في أقصى جنوب الأرض.. ما القصة؟
|
|
|
|
|
|
السيد الصافي يزور قسم التربية والتعليم ويؤكد على دعم العملية التربوية للارتقاء بها
|
|
|
|
لمنتسبي العتبة العباسية قسم التطوير ينظم ورشة عن مهارات الاتصال والتواصل الفعال
|
|
|
|
في جامعة بغداد.. المؤتمر الحسيني الثاني عشر يشهد جلسات بحثية وحوارية
|
|
|
|
للأطفال نصيبهم من جناح جمعية العميد في معرض تونس الدولي للكتاب.. عمّ يبحثون؟
|