آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Complementary DNA |


|
|
Read More
Date: 19-5-2016
Date: 23-2-2021
Date: 5-5-2021
|
Complementary DNA (cDNA(
The base-pair complementarity between a gene and its transcript leads to the formation of a DNA–RNA hybrid under experimental conditions that favor specific hybridization. Pioneering work by Gillespie and Spiegleman that made use of the complementarity between viral genomic DNA and RNA in infected cells (1) helped establish the beginnings of a new technology for the detection of specific RNA molecules hidden among an RNA background, in this case of viral RNA among host cell RNA. The potential advantages of complementary DNA (cDNA) as a probe for RNA detection were recognized before techniques existed to make specific cDNA conveniently for tagging genes. The great abundance in the genome of extra, nontranscribed DNA, which is a common feature of higher organisms, complicates the use of genomic DNA as a cDNA probe. Sometime after the first references to cDNA, a DNA polymerase activity that depends on the presence of an RNA template was observed in RNA viruses (2, 3). This discovery, in addition to extending our knowledge of RNA virus replication intermediates, led to the identification and isolation of a previously unknown form of DNA polymerase called reverse transcriptase and to the identification of buffer conditions that allow some DNA-dependent DNA polymerases to use a DNA-primed RNA template to form a DNA product (4, 5). Largely due to the discovery of reverse transcription and the technical advantages it provides for in vitro cDNA synthesis, cDNA has become a fundamental research tool for use with higher organisms.
1. cDNA Copies of messenger RNA
Transcriptionally active genes represent only a relatively small fraction of the total host genomic DNA in higher organisms, only 3% in human cells, with the noncoding or nonexpressed DNA representing the majority of the genome. Reverse transcribed cDNA synthesized under carefully controlled conditions is a faithful and stable double-strand DNA copy of cellular RNA and is trimmed of excess genomic sequence. A full-length cDNA molecule made from messenger RNA (mRNA) contains only the protein-coding region of an expressed gene, together with the adjacent untranslated regulatory sequences; it lacks any unexpressed gene or genomic sequences not contained in the mature mRNA, such as introns (Fig. 1).
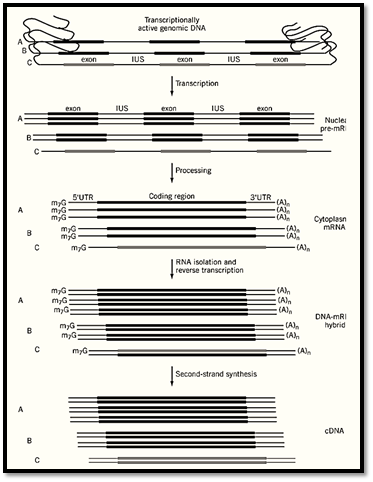
Figure 1. Schematic representation of cDNA synthesis from mRNA. Transcriptionally active genes are transcribed, resulting in pre-mRNA that is processed to the mature form. Nuclear processing includes the removal of intervening sequences (IVS, introns) and addition of a 5′ m7G cap and 3′ poly dA tail. Different gene activities result in different mRNA abundance shown as A, B, or C. After mRNA isolation and reverse transcription, a population of DNA–mRNA hybrid molecules are formed where the DNA is anti-sense in relation to the mRNA. Second-strand synthesis produces a cDNA population in which the more active gene is represented more than the less active gene.
For research that involves mRNA, the preparation of cDNA not only provides a DNA strand complementary to RNA, but it also serves to minimize the technical problems of working directly with RNA, which is chemically unstable at alkaline pH and susceptible to notoriously stable and ubiquitous cellular ribonucleases. A further advantage of cDNA is that in many applications it is integrated into a prokaryotic DNA vector to form recombinant DNA capable of autonomous replication when transformed into a host organism with a favorable genetic background. Engineered vectors with a variety of features are available for many different methods of cDNA analysis. As recombinant DNA, cDNA may be amplified through culturing of the host cells. Because each transformed host cell receives a single recombinant vector, clonal lines of host cells differing only in the recombinant vector received are established. As a copy of total cellular mRNA, however, cDNA is a complex mixture representing the complexity of the template mRNA used to direct its synthesis. Thus, cDNA integrated into a vector, transformed into a host, and cultured represents a complex mixture or library of transcripts, a cDNA library. The use of cDNA frequently requires the selection of a single clone or cDNA copy from a library, since the analysis of expression from a single gene is often the desired aim. The efficient selection of a previously uncharacterized cDNA clone from an entire cDNA library poses a major technical challenge. Selection of a new cDNA is complicated by the lack of any previous information to assist in the selection process. Often, the selection might start with little information other than observations about the encoded function of the desired cDNA clone or atypical cellular characteristics that could be associated with it.
As a stable copy of RNA, a single selected cDNA clone may be transcribed through vector signals to regenerate the RNA, which may in turn be used for synthesis of the encoded protein, either within a host cell or through the use of protein biosynthesis in vitro system. The purity of RNA or protein prepared from a single cDNA clone is absolute, thus justifying the effort to prepare a cDNA library and select a desired clone. This is an alternative to the task of biochemical purification of a protein from a tissue or cell extract. A purified cDNA in a regulated protein expression system may be used to provide a continuous renewable protein source for analysis of the structure, activity, or any characteristic of the encoded protein. Also, the effect that a cDNA's expression has on a relevant eukaryotic cell line may provide information about the biological role of the encoded function. Thus, cDNA-directed protein expression not only provides a source of pure protein for either in vitro or in vivo analyses, but it may also provide a method of cloning (selection) of a cDNA from a cDNA library. Many selections of new cDNAs start without protein or gene sequence information, but instead rely on the detection of the function or biological effect. This process is known as direct expression or functional cloning. Any direct expression cloning method mimics the classic genetic methods of rescue or complementation and so could be considered genetics in the absence of mating. To describe functional cloning in a general way, an altered host cell's phenotype or measured activity is restored to the wild-type when the genetic material encoding the host's deficiency is supplied. Although experimental approaches that rely on classic genetic methods have produced much information about cellular processes, they depend on good genetic systems to which the genetic material can be delivered. Systems that lend themselves to an imposed manipulation and selection, generally single-cell organisms with a short generation time, are best for this purpose. A myriad of eukaryotic cell cultures that have been purposefully altered or carefully isolated from the affected organs of diseased patients and that show an atypical phenotype have been widely used as host cells for functional cloning. The applications for the functional cloning of new cDNAs from higher organisms also depend on a selectable phenotype or activity to isolate the host cell that harbors the relevant genetic material. As an example, the anchorage-dependent phenotype of nearly all primary tissue cells is not exhibited by many tumor cells that will grow in suspension (6). When the genetic material used is cDNA, the details of the selection method must be designed so that the cDNA library contains the cDNA encoding the deficient host activity, and so that the cDNA is incorporated into the host cells and expressed functionally. With expression of a cDNA library in eukaryotic host cells, the selection of a desired clone depends on acquisition of a new phenotype. In the example of the anchorage-independent phenotype exhibited by some tumor cells, the induction of the anchorage dependency allows selection for a cDNA-encoded activity related to the anchorage phenotype and possibly to the cause of tumor formation (7, 8). For any selection where the desire cDNA is of an mRNA that is in low abundance, the selection will require a large-scale transformation of host cells, in order to produce even a single positive clone.
Acquiring a cDNA opens up many types of experimental approaches for the analysis of gene expression and studies of the protein encoded by the cDNA. As an example, protocols are available to target a single codon in a cDNA for site-directed mutagenesis. By using available methods to express the altered cDNA, the effects of the mutation on the encoded protein's activity may be measured, and in this way, a structure–function relation is probed.
Although the methods to prepare and make use of cDNA are advanced and commercially available, it remains a challenge to select a completely new cDNA clone from a cDNA library. This may limit the uses of cDNA technology to better characterize known cellular processes rather than uncharacterized cellular processes. The successful recovery of a novel cDNA depends on a specific selection strategy that ultimately will test each cDNA for an encoded activity. The task of individually testing each clone in a cDNA library is nearly overwhelming. If a screening method designed to test each cDNA in a collective manner cannot be designed, it forces the need for biochemical methods to purify the protein activity in order to provide enough sequence information to obtain the cDNA by more conventional methods, such as library screening with a sequence probe or selective PCR library amplification with degenerate oligonucleotide primers. Evolving methods of protein expression have demonstrated the potential of cDNA selection as a possible alternative to protein purification. However, functional cloning may not always be possible. An alternative approach, which can temporarily bypass the challenge presented by the need to design or apply a new isolation strategy for each new cDNA, is to randomly isolate and analyze cDNA clones first and then subsequently to identify the encoded function. The use of sequence information derived from randomly selected cDNA samples has become part of the Human Genome Project as a way of providing an aid for the physical mapping of expressed genes, as gene tags (9, 10). The cDNA sequences obtained are collected into the expressed sequence tag (EST) database, which is a collection of nonrepetitious human cDNA sequences of randomly selected cDNAs from 250 cDNA libraries from RNA isolated from 37 distinct human organs and tissues (11). The EST information is providing genetic markers for physical gene mapping and also information about the expression pattern of the mapped genes as the cell lineage is known for each sample analyzed. Some hints about the encoded function of the mapped genes may be revealed by the expression pattern. Furthermore,
the large volume of sequence information produced by random sequencing may be compared to the available sequence information of the growing number of fully sequenced genes, in order to identify the presence of conserved protein domains with established activities in a process that has come to be known as functional genomics (12, 13). This process of extending available information about function to randomly acquired new sequence information, which necessarily requires a unified, large-scale approach, will undoubtedly contribute greatly to the identification of the encoded functions of previously unknown human genes.
2. Preparation of cDNA
2.1. Isolation of RNA Template
The preparation of cDNA begins with the isolation of the RNA that will serve as a template to direct the cDNA synthesis. Since cDNA is representative of the RNA template, an inciteful choice of a cellular source of RNA, such as a tissue that is known or at least thought to contain the specific mRNA of interest, is important. In some cases, the RNA might be tested for the presence of an RNA of interest. In a simple case, the RNA sample could be analyzed prior to use for cDNA preparation by use of a sequence probe. This sort of analysis requires, of course, some definite sequence information about the mRNA of interest. In a more complex case, when no sequence information about the mRNA is available, but detection of the encoded protein is possible, the RNA source could be analyzed for the ability to direct synthesis of the protein. Detection of a protein may be accomplished by any known function, such as enzymatic activity or ligand binding activity. The presence of a protein in a cell does not insure the presence of the protein-encoding mRNA, as the protein could be present as a stable form from an earlier expression of an mRNA that is now degraded. On the other hand, a mRNA encoding a protein of interest could be present in a tissue, but remain untranslated. One technique that can be used to insure the presence of the mRNA of interest in an RNA preparation is to translate the total RNA in an in vitro translation reaction and then assay the total resulting protein product for the expected protein. The two high-activity in vitro translation systems in common use are those prepared from rabbit reticulocytes and wheat germ (14, 15).
Given that a tissue source can be identified that contains the RNA of interest, there may be a possibility of enriching for the desired mRNA. Only a small fraction of total cellular RNA is mRNA that is directed to ribosomes for translation into protein (Table 1). The majority of cellular RNAs function in other ways to aid translation, often with structural roles. Enrichment methods aim to remove the more abundant structural RNAs selectively from the mRNAs. Enrichment of polyA + mRNA by hybridization with immobilized oligo (dT), followed by stringent washing to remove the abundant, structural, poly A – RNAs, is a common method used to prepare the mRNA template for synthesis of cDNA representing protein-encoding mRNAs (16). To reduce contamination by mitochondrial poly A + RNA, the mitochondria may be removed by cell fractionation prior to total RNA extraction, although the extra time and handling of cells required could lead to some loss or degradation of RNA. Isolation of full-length mRNA of nuclear origin based on the unique m7G cap structure might also serve as an alternative to cell fractionation to reduce those RNA messages of mitochondrial origin often present in RNA preparations (17). Translational inhibitors have been used to stabilize ribosome-bound mRNA that is degraded during translation (18), and as selective RNA degradation pathways are increasingly well characterized, there is an increasing promise of specific inhibitors of RNA degradation for use in preparing mRNA that normally has a relatively short half-life.
Table 1. Cellular Distribution of RNA in HeLa Cells
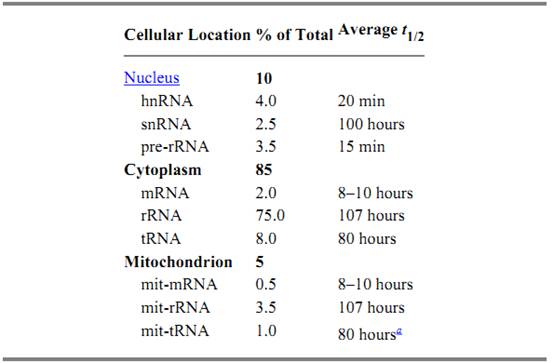
a Note: These values are approximate ones based on experimental observations made during repeated isolation of RNA from HeLa cell cultures.
Among poly A + RNA, there are messages of low, middle, and high abundance. The process of normalizing cDNA is used to reduce the relative number of high- and middle-abundance mRNA represented in the cDNA product. In this way, relatively fewer cDNA clones need to be screened in order to isolate a low-abundance message copy from a cDNA library. Preferred removal of cDNAs representing high- or middle-copy abundance mRNA is based on the rate of strand reannealing in a denatured cDNA sample (19). The two strands of a cDNA representing a high-abundance message have a higher probability of finding each other in a mixture and anneal before those of low-abundance cDNA. When annealed or double-stranded, the cDNA is separated from unannealed or single-strand cDNA by the use of hydroxyapatite chromatography. After a short annealing time, the low-abundance cDNA will remain in the single-strand chromatography fractions. These fractions are collected and allowed to anneal fully for use as normalized cDNA. Alternatively, subtractive hybridization is a technique used for the enrichment of, eg, tissue-specific or developmental stage-specific mRNA in the preparation of subtraction cDNA libraries.
2.2. Synthesis of the cDNA First Strand The two cDNA strands are prepared independently. In the first-strand synthesis, the use of an RNA-dependent DNA polymerase results in a mixture of cDNA–RNA hybrid molecules in which the DNA strand is complementary to the original RNA, but possibly of shorter length (Fig. 2). The reverse transcription systems in common use are based on the avian myeloblastosis reverse transcriptase (AMV-RT) prepared from purified avian myeloblastosis virus particles or on the Moloney murine leukemia virus reverse transcriptase (MMLV-RT) prepared from a recombinant Escherichia coli strain (20). The purified AMV-RT has a stronger endogenous RNAse H activity than the cloned MMLV-RT, thus increasing the possibility of unwanted side reactions when using AMV-RT. However, purified AMV-RT may be more stable than MMLV-RT at the elevated temperatures used to reduce premature termination caused by secondary structure in the RNA template. RNase H is an endoribonuclease that specifically hydrolyzes the phosphodiester bonds of RNA in a DNA–RNA hybrid to produce products with terminal groups 3′-OH and 5′-P. Both AMV-RT and MMLV-RT have a tendency to begin second-strand synthesis prematurely through a reaction in which reverse transcriptase uses DNA rather than RNA as a template by forming a hairpin structure that serves as a template-primer substrate (21). Hairpin formation may be inhibited by the addition of sodium pyrophosphate and spermidine, although the RNase H activity is not inhibited with these conditions. The RNase H activity present in AMV-RT may provide a better opportunity for favorable hairpin formation by leaving single-stranded regions of DNA available for self-hybridization; this side reaction is more of a problem when AMV-RT is used. Heat-stable DNA polymerase isolated from Thermus thermophilus is capable of reverse transcription in the presence of MnCl 2, which is a common alternative to the use of reverse transcriptases (22). Each system of reverse transcription has some advantages, as will be discussed, and should be considered with a view of how the second-strand synthesis will be performed. In addition to reverse transcriptase, first-strand synthesis requires deoxyribonucleotide triphosphates, dATP, dGTP, dTTP, and dCTP (or 5-metyl dCTP which may be substituted for dCTP in order to protect the cDNA from 5-methyl dCTP-sensitive restriction enzymes that may be used in subsequent cloning steps). The first-strand synthesis reaction also requires primers.
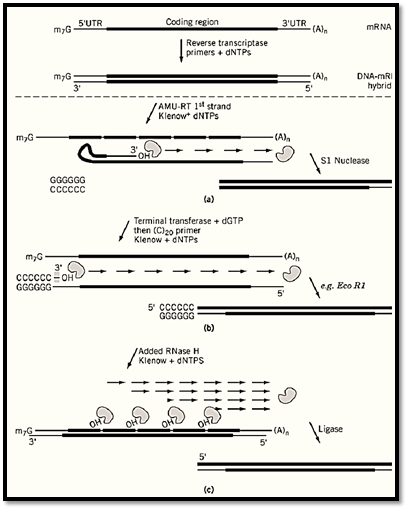
Figure 2. Three separate schemes of second-strand cDNA synthesis from the DNA–mRNA hybrid. (a) A classic method makes advantage of a looped back first strand as a primer for second-strand synthesis. After second-strand synthesis, S1 nuclease digestion of the single-strand loop structure results in an available free end for vector incorporation. (b) Terminal transferase-directed extension of the 3′ end of the first strand results in a dG stretch that provides a binding sight for an oligo dC primer to initiate second-strand synthesis. The undesired dG stretch on the first-strand oligo dT primer is remove by restriction endonuclease digestion targeted to a site present in the oligo dT primer or in the recipient vector if vector priming is used. (c) Added RNase H degrades the RNA in the DNA–RNA hybrid product of first-strand synthesis, resulting a series of RNA primers that are extended during second-strand synthesis. Subsequent ligation may aid the recovery ofull-length cDNAs.
Unlike RNA polymerases, which bind a recognition sequence in a double-strand DNA template to initiate polymerization, replicative-type DNA polymerases require primers with a free 3′-OH group and a short double-stranded sequence. Note that a consideration of the primers to be used for the first-strand synthesis may serve to direct a more representative cDNA or cDNA better suited to the research aim. Because all mRNA, except animal histone mRNA, terminate with a 3′ poly A tract, the use of a p(dT)10–15 oligonucleotide as a first-strand primer ensures that the synthesis begins at the 3′ end of mRNA, even when total RNA is used as a template for reverse transcription. A disadvantage of this primer is that the 5′ end of longer mRNA may be underrepresented in the cDNA product, since the chance that processive synthesis will continue to the 5′ end decreases with product length. The mRNA 3′ terminal poly A tracts with undesirable average lengths of ≈200 nucleotides can be avoided by the use of an “anchored p(dT) oligo,” which has an equal concentration of dA, dC, or dG at the 3′ end of p(dT)12 dN. Such an oligo mixture initiates first-strand synthesis at the position immediately adjacent to the beginning (5′) of the poly A tract, with an expected reduction in the length of the poly A tract captured in the cDNA. A random primer of six oligodeoxynucleotides used in place of p(dT)10–15 may result in a more representative first-strand synthesis, since at least some of the primers should find matching hybridization sequences near the middle and 5′ end of the RNA and thus are frequently extended to the 5′ end of the template. A random primer is a better choice for use with poly A(–)RNA, which is not complementary to p(dT)10–15. A technical consideration for random primed first-strand synthesis is the amount or ratio of primer to template that should be used, since more than one primer for each template could lead to interference between polymerase initiating on different primers hybridized to the same RNA template molecule. This will result in premature termination and a smaller average cDNA product length in the sample. Mixtures of random-primed and oligo dT-primed first-strand synthesis products should equally represent the length of RNA.
Another choice of primer to initiate first-strand synthesis is a sequence-specific primer, a primer complementary to a known sequence within the mRNA of interest. The design of a sequence-specific primer depends, of course, on some available sequence information. A very conserved sequence common to a group of possibly related mRNAs used for the preparation of a limited cDNA library is an example of the application of a sequence-specific primer. Whichever type of primer is used for the initiation of first-strand synthesis, extra sequence at the 5′ end of a primer, such as a restriction site sequence or a second primer-binding site, should not interfere with polymerase-directed extension of the 3′ primer end. Thus, the 5′ end of the primer may serve as a “sticky end” to incorporate the cDNA into a vector or as a primer target for subsequent amplification of the cDNA by PCR.
2.3. Synthesis of the Second Strand
As for first-strand synthesis, second-strand synthesis requires a polymerase, dNTPs, and a primer added to a template. In this case, the template is the product of the first-strand synthesis reaction. Since the template is DNA, a DNA-dependent DNA polymerase, most commonly E. coli DNA polymerase I or the Klenow fragment of DNA polymerase I, is used for second-strand synthesis. Additionally, reverse transcriptase added as a “chaser” reportedly facilitates second-strand synthesis through obstructions caused by secondary structure in the template (23). The product of the second-strand synthesis reaction is double-strand cDNA ready for steps leading to recombinant vector integration. The choice of primers that might serve to direct the synthesis of the second strand is limited, since the 3′ terminal sequence of the first strand varies greatly among the first-strand synthesis product population; there is no common sequence at the 3′ end of the first-strand product available for primer hybridization. Primer design for second-strand synthesis has conventionally been approached in one of three ways: (1) the classic method of hairpin-loop priming followed by S1 nuclease digestion, (2) RNAse H treatment to generate RNA primers, or (3) homopolymeric tailing, also known as vector priming (see Fig. 2). RNAse H is commonly available in a pure form for the second-strand synthesis reaction (Fig. 2c). Added RNAse H is used to generate short RNA primers that are extended to form the second strand (24, 25). The relative technical simplicity, high yield, and large average product length achieved with the RNAse H method have proven it to be the superior approach generally, although other methods may continue to have specific applications and certainly have had a central role in characterization of the potential side reactions that affect the yield and average size of the cDNA product.
The classic method of second-strand synthesis (26, 27) takes advantage of the tendency for the 3′ end of the first strand to fold back and form a DNA duplex with sequences located further 5′ or upstream on the same DNA strand (Fig. 2a). A consequence of the duplex or hairpin formation is availability of the self-primed template for second-strand synthesis. After removal of the RNA, reagents for second-strand synthesis are added, and so the second strand is generated. The product is double-strand cDNA covalently closed at the end corresponding to the 5′ end of the original mRNA. The addition of S1 nuclease, a single-strand-specific nuclease, results in cleavage of the single-strand loop to produce free 5′ and 3′ ends, which are necessary for subsequent integration into a cloning vector. Although the use of S1 nuclease, which has a potential of hydrolyze double-stranded DNA when used in excess, is often cited as the major disadvantage of the classical method, it is the necessary loss of sequence information that is the true technical fault in the method. Since at least some sequence is involved in the hairpin formation, the sequence of the 5′ end of the original mRNA is not represented in the cDNA product when this classical method of second-strand synthesis is used.
The method of homopolymeric tailing or vector priming relies on the use of terminal deoxynucleotidyltransferase, an enzyme that catalyzes the sequential addition of available nucleotide triphosphates to the free 3′ end of a DNA strand. Thus, a homopolymer tail made by extension of the free 3′ end of the cDNA strand resulting from the first-strand synthesis reaction serves as the primer site for second-strand synthesis (Fig. 2b). The application of homopolymer tailing to second-strand synthesis preserves the cDNA sequence representing the original 5′ end of the mRNA template, since priming is from sequence added by terminal transferase (28, 25). After first-strand synthesis initiated from a T-tailed vector, the 3′ end of the newly formed cDNA strand is the target for homopolymer tailing. The use of dGTP for the tailing reaction consistently results in an average of 20 G nucleotides on both the 3′ end of the cDNA first strand and on the free 3′ end of the vector. The consistent length of the tail with use of dGTP is apparently due to structural constraints, which cause decreased tailing after 20 Gs (29, 30). A restriction site in the vector is used to remove the G-tail from the vector, without affecting the G-tail on the cDNA, which as a DNA–RNA hybrid is a poor substrate for restriction endonucleolytic cleavage (31). Finally, an oligo p(dC)20 primer is added, along with other reagents required for second-strand synthesis. The p(dC)20 primer may be designed to have a 5′ end complementary to the overhang of the vector restricted end to aid in subsequent circularization of the recombinant DNA; otherwise, both the cDNA and vector ends may be made flush or blunt prior to circularization. Many variations of the vector priming method briefly described here have been described in detail elsewhere, but all make use of terminal transferase (32.( The disadvantages of being more technically difficult and having more steps, as well as having a stretch of 20 G nucleotides at the vector-cDNA boundary (which could require special techniques for sequencing and subcloning), might be balanced by the important potential of increasing the number of full-length clones when using this method of second-strand synthesis.
Isolation of a cDNA corresponding to the intact full-length mRNA molecule often presents a challenge. Methods that increase the frequency of obtaining full-length clones in a cDNA synthesis reaction are often preferred. Losses of cDNA may occur at each step of the synthesis, from RNA isolation to vector incorporation. Losses of full-length cDNA may also occur due to incomplete replication of template by polymerase. For example, the presence of secondary structure in the RNA template may impede the progress of reverse transcriptase during first-strand synthesis, resulting in the formation of a partial cDNA beginning at the primer site and ending at the site where the secondary structure is encountered by the polymerase. Technical manuals that contain detailed protocols for cDNA synthesis often describe ways to monitor the progress of cDNA synthesis to give an indication of the reaction yields, or the incurred losses, and also the average size of the cDNA strand. A method designed to extend a partial cDNA isolated by a selection procedure called RACE (rapid amplification of cDNA ends) may be used to obtain the corresponding full-length clone (33). The actual 5′ end of an mRNA should, however, be mapped against the gene and/or correlated with the N-terminal sequence of the protein where possible. Primer Extension is frequently used to determine the precise 5′ end of an mRNA, and this method only requires enough sequence information for primer design.
3. Uses of cDNA
The final steps in cDNA synthesis are often those leading to vector integration. The use or application of cDNA is largely determined by the type of vector that is chosen to contain the cDNA. Vector integration is, therefore, the first step toward fitting the cDNA to the type of application that will be made or toward developing a selection strategy for the isolation of a cDNA of interest from a library. Recent progress in the development of cloning vectors for specialized applications and of methods of efficiently adapting the cDNA ends to fit the vector ends have resulted in a lot of flexibility in the combination of available vector features when making a choice from the long list of commercially available cloning vectors. The immediate aim of retaining all the newly synthesized cDNA through the process of vector integration and host transformation, while keeping the desired vector features, is frequently satisfied by the use of a phagemid (34). A phagemid vector combines features of lambda phage, filamentous phage, and plasmid vectors into one cloning vector (35, 36.( The efficiency of host cell transformation through packaging of cDNA into viral particles, and subsequent viral infection as performed with the use of a l phage cloning vector, cannot be matched by even the optimal conditions for direct host-cell transformation with plasmid DNA. There is consequently less potential for cDNA loss during host cell transformation with a l phage system when compared to a plasmid system. The lower background that results from the screening of plaques rather than colonies with sequence hybridization probes or antibody probes offers another significant advantage to the use of viral vectors. Plasmid vectors offer advantages over l phage vectors for the characterization of the cDNA inserts, due to the necessarily large size of l phage vectors. Phagemid vectors contain a plasmid vector within a l phage vector and additional sequences derived from filamentous phage that contribute to the in vivo excision of the plasmid from the l phage vector. Thus, for characterization of individual selected cDNA clones, the original phage clones are converted to plasmid clones. Although it is technically straightforward to subclone a single cDNA clone from one vector to another, it is technically difficult to subclone an entire cDNA library from one to another vector without some loss of cDNA. Whichever type of vector is used to receive newly made cDNA, consideration of the vector features required for selection of a cDNA of interest from a library and the efficiency of the process of integration and host-cell transformation should be given prior to vector integration of cDNA as a library.
Possibly the greatest benefit of aquiring a new cDNA of interest from a cDNA library is the sequence information that can be derived from it. In addition to the encoded protein sequence, comparison of a cDNA sequence with the corresponding gene sequence reveals or confirms the location of any introns present in the gene. Analysis of a set of positives identified by rescreening of a library with the now available hybridization probe may provide some indication of the variability of the gene transcripts, eg, the presence of alternate splice sites or multicopy genes with minor sequence variations. The presence of viral RNA polymerase recognition sequences, most commonly SP6 and T7 promoters, in many cloning vectors are useful for the in vitro production of anti-sense RNA probes that may be used for the high-sensitivity detection of gene expression. Likewise, PCR primers may be designed from the sequence information to provide an alternate method of sensitive or quantitative gene expression analysis. The presence of potential post-transcriptional regulatory elements in the untranslated regions represented in the cDNA may be identified through computer-assisted sequence analysis and database comparisons. Although any number of extremely sophisticated uses for cDNA sequence information may be envisioned for a particular application, a generally useful benefit is the possibility to generate peptide antibodies to encoded peptide epitopes predicted to be on the protein surface.
Vectors are available with many combinations of features. A particularly useful vector feature for many types of applications is the inclusion of sequences that support cDNA expression. As the sequences required for correct initiation of translation differ among organisms, expression vectors, or an expression cassette within a cloning vector, are designed for use as a set with a matching host cell. The promoter sequences included to drive the transcription of the expression cassette and efficient transcriptional termination signals are likewise active in the same host. As there are no introns to be removed, expression of many encoded eukaryotic proteins is supported in strains of E. coli, provided that these proteins are not toxic to the E. coli host strain used. In order to avoid the possible toxic effects of some eukaryotic proteins, expression vectors with inducible promoters are available to postpone the cDNA expression to a point of vigorous growth so that significant protein production is accomplished before the host culture dies. The activity of eukaryotic proteins produced in prokaryotic host cells may be deficient due to the absence of factors required for proper protein folding in vivo, for post-translational modifications essential for the activity, or even for correct intracellular localization. Eukaryotic expression systems may better support the production of eukaryotic proteins with natural levels of activity and are designed for maximum protein production. Such expression systems may support continued replication of the vector containing a cDNA insert, in addition to containing an expression cassette with an active promoter and signals for transcription termination and translation initiation. Both prokaryotic and eukaryotic expression systems with matched vector and host are designed to produce proteins in quantities required for their structural characterization from manageable volumes of cell culture. Accurate physical measurements that require purified proteins may take advantage of “built-in” purification aids available in some expression vectors. Expression of a cDNA as a fusion protein aids in the isolation of protein from cell cultures. Provided a cDNA is cloned in-frame in relation to a vector sequence that encodes a polypeptide chain with a known ligand affinity, the resulting fusion protein may be purified from cell lysates by passage through an affinity chromatography column containing the immobilized ligand. The presence of a proteinase peptidase recognition sequence facilitates proteolytic removal of the vector-encoded polypeptide and elution of the cDNA-encoded protein from the affinity column.
An additional type of expression vector that is adapted to cDNA selection is one designed for use with a yeast host strain. The yeast 1, 2, and 3-hybrid systems have been designed to isolate a cDNA clone from a library based on the encoded protein's recognition or specific binding to a DNA, protein, or RNA sequence, respectively (37-39). In this way, cDNA selection is facilitated by its expression inside a yeast host cell that has been genetically altered for the selection. In order to perform the selection, an entire cDNA library is cloned into a shuttle vector that will support replication in E. coli, as well as replication and expression (as a fusion protein) in the yeast host. The library is amplified for transformation into the recipient yeast host strain, which is altered in such a way that specific binding of a target sequence or “bait sequence” results in a positive selection. The yeast hybrid systems typify advances in the use of traditional genetic strains adapted for functional cloning of cDNA from higher organisms, and yeast has consequently become a tool for functional cloning through molecular recognition, extending the use of cDNA as an aid to the selection of novel factors. Thus, a factor that binds to a DNA sequence, such as a transcription factor that binds to a gene promoter element, may be difficult to isolate by biochemical means due to its low intracellular abundance, but it might be cloned with the yeast 1-hybrid system. Likewise, a factor that binds to a protein might be cloned with the 2-hybrid system, or an RNA-binding protein with the yeast 3-hybrid system.
4.Summary
cDNA is a faithful double-stranded DNA copy of RNA and, as such, can represent those genes that are expressed as RNA in a source tissue. When integrated into an appropriate cloning vector to form recombinant DNA, cDNA may provide a continuous renewable source of genetic material for hybridization probes, designed reporters of biological activity, or pure encoded protein. The benefits of protein production and a myriad of specific research applications have made cDNA a universal tool for basic and applied cell and molecular biology research. The potential for using cDNA in molecular methods designed to select for activities that contribute detectable cellular phenotypes has provided possible alternatives or improvements to genetic and biochemical approaches.
References
1. D. Gillespie and S. Spiegleman (1965) J. Mol. Biol. 12, 829–842.
2. H. M. Temin and S. Mizutani (1970) Nature 226, 1211–1213.
3. D. Baltimore (1970) Nature 226, 1209–1211.
4. M. G. Sarngadharan et al. (1972) Nature New Biol. 240, 67–72.
5. E. M. Scolnick (1971) Develop. Biol. 26, 175–176.
6. J. C. Barrett et al. (1979) Cancer Res. 39, 1504–1510.
7. M. Noda (1990) Molec. Carcinogenesis 3, 251–253.
8. C. P. Carstens et al. (1995) Gene 164, 195–202.
9. L. Rowen et al. (1997) Science 278, 605–607.
10. M. Boguski and G. D. Schuler (1995) Nature Genet. 10, 36971.
11. M. D. Adams et al. (1995) Nature 377, Suppl., 3–174.
12. S. Henikoff et al. (1997) Science 278, 609–614.
13. R. L. Tatusov et al. (1997) Science 278, 631–637.
14. B. E. Roberts and B. M. Paterson (1973) Proc. Natl. Acad. Sci. USA 70, 2330.
15. C. W. Anderson et al. (1983) Methods Enzymol. 101, 635.
16. H. Aviv and P. Leder (1972) Proc. Natl. Acad. Sci. USA 69, 1408–1412.
17. I. Edery et al. (1995) Mol. Cell Biol. 15, 3363–3371.
18. J. Ross (1997) Bioessays 19, 527–529.
19. T. G. Coche (1997) Methods Mol. Biol. 67, 359–369.
20. M. Roth et al. (1985) J. Biol. Chem. 260, 9326–9335.
21. M. S. Krug and S. L. Berger (1987) Methods Enzymol. 152, 318.
22. C. Rüttimann et al. (1985) Eur. J. Biochem. 149, 41–46.
23. U. Gubler (1987) Methods Enzymol. 152, 325.
24. H. Okayama and P. Berg (1982) Mol. Cell Biol. 2, 161–170.
25. U. Gubler and B. J. Hoffman (1983) Gene 25, 263–269.
26. A. Efstratiadis et al. (1976) Cell 7, 279–288.
27. F. Rougeon and B. Mach (1976) Proc. Natl. Acad. Sci. USA 73, 3418–3422.
28. H. Land et al. (1981) Nucl. Acids Res. 9, 2251–2266.
29. A. Dugaiczyk et al. (1980) Biochemistry 19, 5869–5873.
30. A. Otsuka (1981) Gene 13, 339–346.
31. W. H. Eschenfeldt et al. (1987) Methods Enzymol. 152, 339.
32. P. L. Deininger (1987) Methods Enzymol. 152, 376.
33. M. A. Frohman et al. (1988) Proc. Natl. Acad. Sci. USA 85, 8998–9002.
34. M. A. Alting-Mees et al. (1992) Methods Enzymol. 216, 483.
35. D. Hanahan (1983) J. Mol. Biol. 166, 557–580.
36. J. M. Short et al. (1988) Nucl. Acids Res. 16, 7583–7600.
37. S. Fields and O. Song (1989) Nature 340, 245–247.
38. D. J. SenGupta et al. (1996) Proc. Natl. Acad. Sci. USA 93, 8496–8501.
39. Z. F. Wang et al. (1996) Genes Develop. 10, 3028–3040.

|
|
|
|
اكتشاف تأثير صحي مزدوج لتلوث الهواء على البالغين في منتصف العمر
|
|
|
|
|
|
|
زهور برية شائعة لتر ميم الأعصاب التالفة
|
|
|
|
|
|
بوقت قياسي وبواقع عمل (24)ساعة يوميا.. مطبعة تابعة للعتبة الحسينية تسلّم وزارة التربية دفعة جديدة من المناهج الدراسية
|
|
|
|
يعد الاول من نوعه على مستوى الجامعات العراقية.. جامعة وارث الانبياء (ع) تطلق مشروع اعداد و اختيار سفراء الجامعة من الطلبة
|
|
|
|
قسم الشؤون الفكرية والثقافية يعلن عن رفد مكتبة الإمام الحسين (ع) وفروعها باحدث الكتب والاصدارات الجديدة
|
|
|
|
بالفيديو: بمشاركة عدد من رؤساء الاقسام.. قسم تطوير الموارد البشرية في العتبة الحسينية يقيم ورشة عمل لمناقشة خطط (2024- 2025)
|