آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Gene Knockouts, Transgenics, and Genome Editing |


|
|
Read More
Date: 2-4-2021
Date: 11-5-2016
Date: 28-12-2015
|
Gene Knockouts, Transgenics, and Genome Editing
KEY CONCEPTS
-Embryonic stem (ES) cells that are injected into a mouse blastocyst generate descendant cells that become part of a chimeric adult mouse.
-When the ES cells contribute to the germline, the next generation of mice can be derived from the ES cell.
-Genes can be added to the mouse germline by transfecting them into ES cells before the cells are added to the blastocyst.
-An endogenous gene can be replaced by a transfected gene using homologous recombination.
-The occurrence of successful homologous recombination can be detected by using two selectable markers, one of which is incorporated with the integrated gene, the other of which is lost when recombination occurs.
-The Cre/lox system is widely used to make inducible knockouts and knock-ins.
-Several tools exist to edit the genome directly in living cells.
An organism that gains new genetic information from the addition of foreign DNA is described as transgenic. For simple organisms such as bacteria or yeast, it is easy to generate transgenics by transformation with DNA constructs containing sequences of interest. Transgenesis in multicellular organisms, however, can be much more challenging.
The approach of directly injecting DNA can be used with mouse eggs, as shown in FIGURE 1. Plasmids carrying the gene of interest are injected into the nucleus of the oocyte or into the pronucleus of the fertilized egg. The egg is implanted into a pseudopregnant mouse (a mouse that has mated with a vasectomized male to trigger a receptive state). After birth, the recipient mouse can be examined to see whether it has gained the foreign DNA, and, if so, whether it is expressed. Typically, a minority (~15%) of the injected mice carry the transfected sequence. In general, multiple copies of the plasmid appear to have been integrated in a tandem array into a single chromosomal site. The number of copies varies from 1 to 150, and they are inherited by the progeny of the injected mouse. The level of gene expression from transgenes introduced in this way is highly variable, both due to copy number and the site of integration. A gene can be highly expressed if it integrates within an active chromatin domain, but not if it integrates in or near a silenced region of the chromosome.
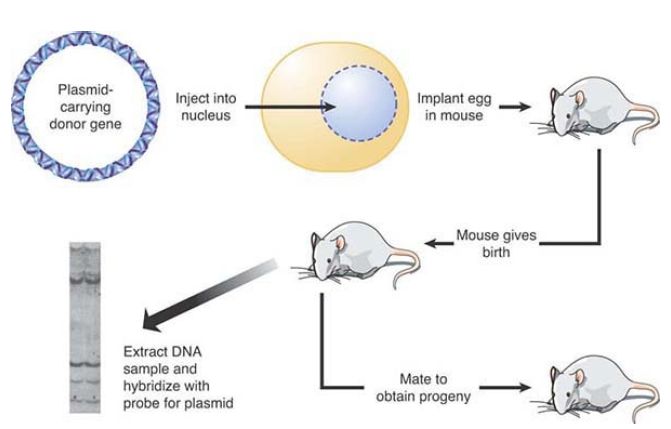
FIGURE 1. Transfection can introduce DNA directly into the germline of animals.
Photo reproduced from: Chambon, P. 1981. Sci Am 244:60–71. Used with permission of Pierre Chambon, Institute of Genetics and Molecular and Cellular Biology, College of France.
Transgenesis with novel or mutated genes can be used to study genes of interest in the whole animal. In addition, defective genes can be replaced by functional genes using transgenic techniques.
One example is the cure of the defect in the hypogonadal mouse. The hpg mouse has a deletion that removes the distal part of the gene coding for the precursor to gonadotropin-releasing hormone (GnRH) and GnRH-associated peptide (GAP). As a result, the mouse is infertile. When an intact hpg gene is introduced into the mouse by transgenic techniques, it is expressed in the appropriate tissues. FIGURE 2. summarizes experiments to introduce a transgene into a line of hpg–homozygous mutant mice. The resulting progeny are normal. This provides a striking demonstration that expression of a transgene under normal regulatory control can be indistinguishable from the behavior of the normal allele.
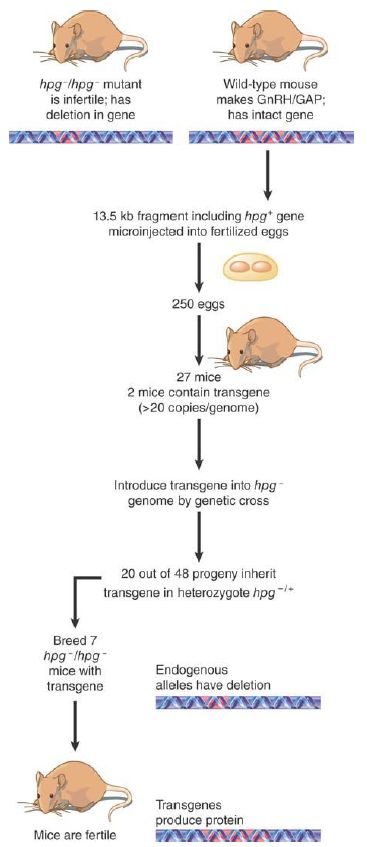
FIGURE 2. Hypogonadism can be averted in the progeny of hpg mice by introducing a transgene that has the wild-type sequence.
Although promising, there are impediments to using such techniques to cure human genetic defects. The transgene must be introduced into the germline of the preceding generation, the ability to express a transgene is not predictable, and an adequate level of expression of a transgene can be obtained in only a small minority of the transgenic individuals. In addition, the large number of transgenes that might be introduced into the germline, and their erratic expression, could pose problems in cases in which overexpression of the transgene is harmful. In other cases, the transgene can integrate near an oncogene and activate it, promoting carcinogenesis.
A more versatile approach for studying the functions of genes is to eliminate the gene of interest. Transgenesis methods allow DNA to be added to cells or animals, but to understand the function of a gene, it is most useful to be able to remove the gene or its function and observe the resulting phenotype. The most powerful techniques for changing the genome use gene targeting to delete or replace genes by homologous recombination. Gene deletions are usually referred to as knockouts, whereas replacement of a gene with an alternative mutated version is called a knock-in.
In simple organisms such as yeast, this is again a very simple process in which DNA encoding a selectable marker flanked by short regions of homology to a target gene is transformed into the yeast. As little as 40 bp or so of homology will result in extremely efficient replacement of the target gene by the introduced marker gene, via homologous recombination using the short regions of homology.
In some organisms, and in mammalian cells in culture, there is no good method for deleting endogenous genes. Instead, researchers use knockdown approaches, which reduce the amount of a gene product (RNA or protein) produced, even while the endogenous gene is intact. There are several different knockdown methods, but one of the most powerful is the use of RNA interference (RNAi) to selectively target specific mRNAs for destruction. Briefly, introduction of double-stranded RNA into most eukaryotic cells triggers a response in which these RNAs are cleaved by a nuclease called Dicer into 21 bp dsRNA fragments (siRNAs), unwound into single strands, and then used by another enzyme, RISC, to find and anneal to mRNAs containing complementary sequence. When a fully complementary mRNA is found, it is cleaved and destroyed. In practice, this means that the mRNA for any gene can be targeted for destruction by introduction of a dsRNA designed to anneal to the target of interest. The means of introducing the dsRNA depends on the species being targeted; in mammalian cells, one method is transfection with DNA encoding a self-annealing RNA that forms a hairpin containing the targeting sequence. For many species, researchers are developing siRNA libraries that allow systematic elimination of large sets of target mRNAs, one at a time, providing a powerful new tool for genetic screening.
In some multicellular organisms, gene deletion is possible, but the process is more complicated than in organisms like yeast. In mammals, the target is usually the genome of an ES cell, which is then used to generate a mouse with the knockout. ES cells are derived from the mouse blastocyst (an early stage of development, which precedes implantation of the egg in the uterus). FIGURE 3. illustrates the general approach.
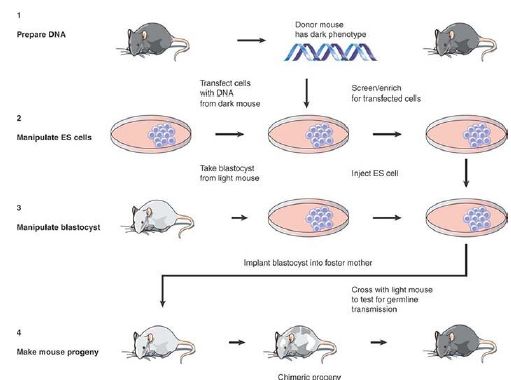
FIGURE 3. ES cells can be used to generate mouse chimeras, which breed true for the transfected DNA when the ES cell contributes to the germline.
ES cells are transfected with DNA in the usual way (most often by microinjection or electroporation). By using a donor that carries an additional sequence, such as a drug-resistance marker or some particular enzyme, it is possible to select ES cells that have obtained an integrated transgene carrying any particular donor trait. This results in a population of ES cells in which there is a high proportion carrying the marker.
These ES cells are then injected into a recipient blastocyst. The ability of the ES cells to participate in normal development of the blastocyst forms the basis of the technique. The blastocyst is implanted into a foster mother, and in due course develops into a chimeric mouse. Some of the tissues of the chimeric mice are derived from the cells of the recipient blastocyst; other tissues are derived from the injected ES cells. The proportion of tissues in the adult mouse that are derived from cells in the recipient blastocyst and from injected ES cells varies widely in individual progeny; if a visible marker (e.g., coat-color gene) is used, areas of tissue representing each type of cell can be seen. To determine whether the ES cells contributed to the germline, the chimeric mouse is crossed with a mouse that lacks the donor trait.
Any progeny that have the trait must be derived from germ cells that have descended from the injected ES cells. By this means, it is known that an entire mouse has been generated from an original ES cell!
When a donor DNA is introduced into the cell, it might insert into the genome by either nonhomologous or homologous recombination. Homologous recombination is relatively rare, probably representing <1% of all recombination events, and thus occurring at a frequency of ~10-7 . By designing the donor DNA appropriately, though, we can use selective techniques to identify those cells in which homologous recombination has occurred.
FIGURE 4. illustrates the knockout technique that is used to disrupt endogenous genes. The basis for the technique is the design of a knockout construct with two different markers that will allow nonhomologous and homologous recombination events in the ES cells to be distinguished. The donor DNA is homologous to a target gene, but has two key modifications. First, the gene is inactivated by interrupting or replacing an exon with a gene encoding a selectable marker (most often the neoR gene that confers resistance to the drug G418 is used). Second, a counterselectable marker (a gene that can be selected against) is added on one side of the gene; for example, the thymidine kinase (TK) gene of the herpes simplex virus.
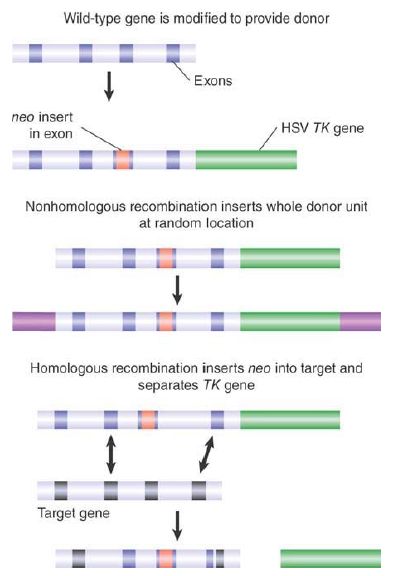
FIGURE 4. A transgene containing neo within an exon and TK downstream can be selected by resistance to G418 and loss of TK activity.
When this knockout construct is introduced into an ES cell, homologous and nonhomologous recombinations will result in different outcomes. Nonhomologous recombination inserts the entire construct, including the flanking TK gene. These cells are resistant to neomycin, and they also express thymidine kinase, which makes them sensitive to the drug ganciclovir (thymidine kinase phosphorylates ganciclovir, which converts it to a toxic product). In contrast, homologous recombination involves two exchanges within the sequence of the donor gene, resulting in the loss of the flanking TK gene. Cells in which homologous recombination has occurred therefore gain neomycin resistance in the same way as cells that have nonhomologous recombination, but they do not have thymidine kinase activity, and so are resistant to ganciclovir. Thus, plating the cells in the presence of neomycin plus ganciclovir specifically selects those in which homologous recombination has replaced the endogenous gene with the donor gene.
The presence of the neoR gene in an exon of the donor gene disrupts translation, and thereby creates a null allele. A particular target gene can therefore be knocked out by this means; once a mouse with one null allele has been obtained, it can be bred to generate the homozygote. This is a powerful technique for investigating whether a particular gene is essential, and what functions in the animal are perturbed by its loss. Sometimes phenotypes can even be observed in the heterozygote.
A major extension of ability to manipulate a target genome has been made possible by using the phage Cre/lox system to engineer site-specific recombination in a eukaryotic cell. The Cre enzyme catalyzes a site-specific recombination reaction between two lox sites, which are identical 34-bp sequences. FIGURE 5 shows that the consequence of the reaction is to excise the stretch of DNA between the two lox sites.
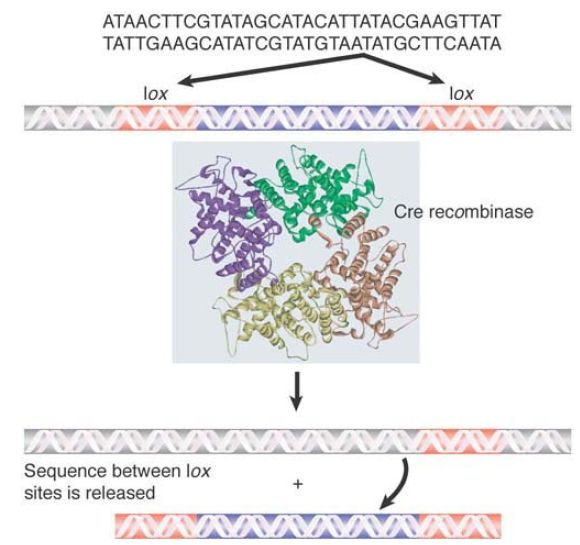
FIGURE 5 The Cre recombinase catalyzes a site-specific recombination between two identical lox sites, releasing the DNA between them.
Structure from Protein Data Bank: 1OUQ. E. Ennifar, et al. 2003. Nucleic Acids Res 31:5449–5460.
The great utility of the Cre/lox system is that it requires no additional components and works when the Cre enzyme is produced in any cell that has a pair of lox sites. FIGURE 6. shows that we can control the reaction to make it work in a particular cell by placing the cre gene under the control of a regulated promoter. The procedure begins with two mice. One mouse has the cre gene, typically controlled by a promoter that can be turned on specifically in a certain cell or under certain conditions. The other mouse has a target sequence flanked by lox sites. When we cross the two mice, the progeny have both elements of the system; the system can be turned on by controlling the promoter of the cre gene. This allows the sequence between the lox sites to be excised in a controlled way.
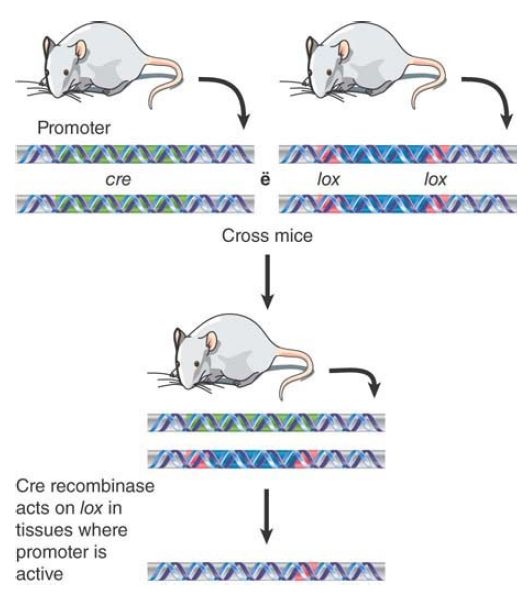
FIGURE 6. By placing the Cre recombinase under the control of a regulated promoter, it is possible to activate the excision system only in specific cells. One mouse is created that has a promotercre construct, and another that has a target sequence flanked by lox sites. The mice are crossed to generate progeny that have both constructs. Then excision of the target sequence can be triggered by activating the promoter.
The Cre/lox system can be combined with the knockout technology to give us even more control over the genome. Inducible knockouts can be made by flanking the neo gene (or any other gene that is used similarly in a selective procedure) with lox sites. After the knockout has been made, the target gene can be reactivated by causing Cre to excise the neoR gene in some particular circumstance (such as in a specific tissue). FIGURE 7. shows a modification of this procedure that allows a knock-in to be created. Basically, we use a construct in which some mutant version of the target gene is used to replace the endogenous gene, relying on the usual selective procedures. Then, when the inserted gene is reactivated by excising the neoR sequence, we have in effect replaced the original gene with a different version.
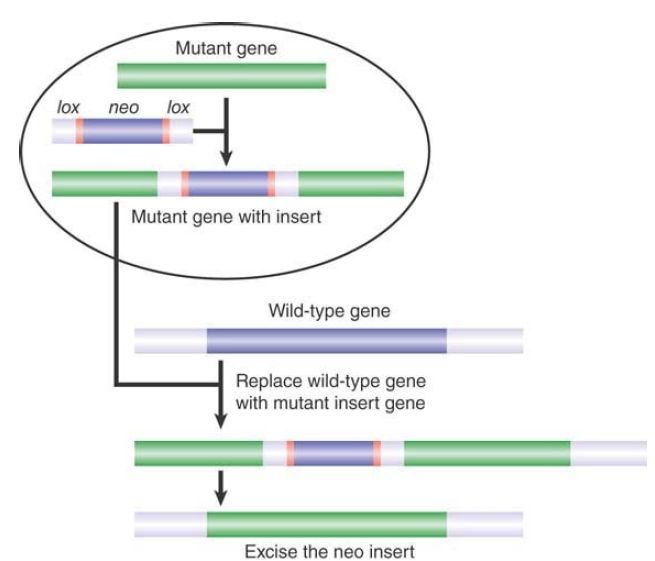
FIGURE 7.An endogenous gene is replaced in the same way as when a knockout is made , but the neomycin gene is flanked by lox sites. After the gene replacement has been made using the selective procedure, the neomycin gene can be removed by activating Cre, leaving an active insert.
A useful variant of this method is to introduce a wild-type copy of the gene of interest in which the gene itself (or one of its exons) is flanked by lox sites. This results in a normal animal that can be crossed to a mouse containing Cre under control of a tissuespecific or otherwise regulated promoter. The offspring of this cross are conditional knockouts, in which the function of the gene is lost only in cells that express Cre. This is particularly useful for studying genes that are essential for embryonic development; genes in this class would be lethal in homozygous embryos and thus are very difficult to study.
Recently, several technologies have emerged that allow direct editing of target sequences in the genome in vivo. These methods are all based on endonucleases that can be targeted very specifically to genomic sites. The double-strand breaks created by these nucleases then utilize the cell’s own repair machinery to generate sequence alterations. These changes can include gene mutation, deletion, insertion, or even precise gene editing or correction based on a provided donor template.
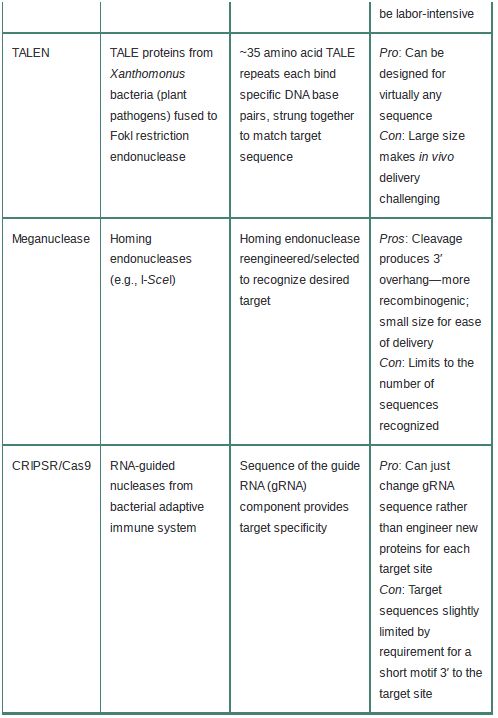
The specificity and outcomes of these techniques depend on the specific targeting of endonucleases to only the site(s) of interest.Four general classes of nucleases are used: zinc finger nucleases (ZFNs), meganucleases, transcription activator-like effector nucleases (TALENs), and, most recently, the CRISPR/Cas9 system. The basic characteristics of these systems are summarized in TABLE 1.
TABLE 1 Basic features of endonuclease-based genome-editing systems.
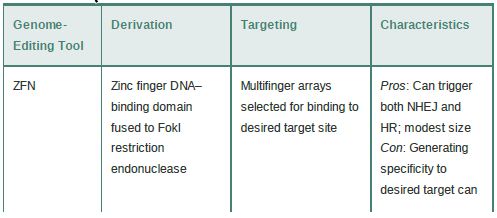
ZFNs take advantage of the fact that zinc finger (ZF) DNA binding domains (discussed in the chapter titled Eukaryotic Transcription) are modular domains that each recognize a 3-bp sequence and can be strung together into multifinger domains to recognize longer sequences. A combination of engineering and selection allows the creation of ZF arrays that will target a locus of interest. The ZF portion is fused to the endonuclease domain of the FokI restriction enzyme to create the ZFN, which then dimerizes to make a DSB at the desired site.
Similarly, TALENs utilize a modular DNA binding repeat; in this case, a set of conserved 33–35 amino acid repeats derived from the TALE proteins of the Xanthomonas bacterial plant pathogens. Each TALE repeat recognizes a single base pair (determined by two variable amino acids within the 33–35 aa repeat), so multiple TALE repeats can be strung together to recognize virtually any sequence (with the only requirement that there be a T at the 5′ end of the target). As for ZFNs, the TALE array is fused to the FokI enzyme to provide the cleavage. A downside of TALENs is that because each base pair in the target site is recognized by an approximately 35 aa motif, targeting sequences long enough to be unique in the genome can result in very large TALENs, which makes delivery into target cells or tissues more challenging.
The meganucleases, despite their name, are actually the smallest of these editing nucleases and thus the easiest to deliver (in fact,several meganucleases with different specificities could be delivered simultaneously for multiplex editing). These nucleases are derived from naturally occurring homing endonucleases, a family of nucleases encoded within introns or as self-splicing inteins. These nucleases naturally recognize long, usually asymmetric, sites of up to 40 bp that typically occur only 1 or 2 times in a genome. (The large target sites are the origin of the name.) Meganucleases can be engineered or selected to recognize novel sequences, but because they lack the modular nature of ZFNs and TALENs, this can be difficult.
The most recent—and most exciting—gene editing tool to be developed is based on the CRISPR-Cas RNA-guided nucleases that form the basis of a bacterial adaptive immune response against viruses and plasmids. The CRISPR-Cas system is described in more detail in the chapter titled Regulatory RNA. Briefly, the CRISPR-Cas system involves integration of invading nucleic acids into CRISPR loci, where they are transcribed into CRISPR RNAs (crRNAs). These then form a complex with a transactivating crRNA and Cas (CRISPR-associated) proteins. The crRNA then targets cleavage of complementary DNA sequences.
To adapt this system for gene editing, the two RNAs are fused into a single guide RNA (gRNA), and changes to a portion of this sequence can be used to define desired targets. This is an enormous advantage over the other technologies, which need to engineer novel proteins for every desired target sequence. The same Cas9 protein can simply be delivered with a gRNA (or
several!) designed against the site of interest. Cas9 proteins do require a short (about 3 bp) protospacer-adjacent motif (PAM) 3′ to the target site, which can limit some target sequences. Recent efforts have focused on developing Cas9 proteins with different PAM specificities to expand this repertoire as well as developing Cas9 variants with increased specificity to reduce off-target cleavage.
With these techniques, we are able to investigate the functions and regulatory features of genes in whole animals. The ability to introduce DNA into the genome allows us to make changes in it, add new genes that have had particular modifications introduced in vitro, or inactivate existing genes. Thus, it becomes possible to delineate the features responsible for tissue-specific gene expression. Gene editing techniques have already begun to show promise as a gene therapy tool to treat human genetic disorders and other diseases. For example, ZFNs have been used in Phase 1 clinical trials to modify the CCR5 receptor (used by HIV to enter cells) in HIV-infected patients. All of the gene editing tools are being utilized in preclinical studies. Ultimately, we can expect routinely to replace or repair defective genes in the genome in a
targeted manner.

|
|
|
|
"عادة ليلية" قد تكون المفتاح للوقاية من الخرف
|
|
|
|
|
|
|
ممتص الصدمات: طريقة عمله وأهميته وأبرز علامات تلفه
|
|
|
|
|
|
|
ندوات وأنشطة قرآنية مختلفة يقيمها المجمَع العلمي في محافظتي النجف وكربلاء
|
|
|