آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Phospholipid Degradation |


|
|
Read More
Date: 25-12-2021
Date: 6-10-2021
Date: 21-11-2021
|
Phospholipid Degradation
The degradation of phosphoglycerides is performed by phospholipases found in all tissues and pancreatic juice. [Note: For a discussion of phospholipid digestion A number of toxins and venoms have phospholipase activity, and several pathogenic bacteria produce phospholipases that dissolve cell membranes and allow the spread of infection. Sphingomyelin is degraded by the lysosomal phospholipase, sphingomyelinase (see B. below).
A. Phosphoglycerides
Phospholipases hydrolyze the phosphodiester bonds of phosphoglycerides, with each enzyme cleaving the phospholipid at a specific site. The major phospholipases are shown in Figure 1. [Note: Removal of the FA from carbon 1 or 2 of a phosphoglyceride produces a lysophosphoglyceride, which is the substrate for lysophospholipases.] Phospholipases release molecules that can serve as second messengers (for example, DAG and IP3) or that are the substrates for synthesis of messengers (for example, arachidonic acid). Phospholipases are responsible not only for degrading phospholipids but also for remodeling them. For example, phospholipases A1 and A2 remove specific FA from membrane-bound phospholipids, which can be replaced with different FA using fatty acyl CoA transferase. This mechanism is used as one way to create the unique lung surfactant DPCC (see p. 204) and to insure that carbon 2 of PI (and sometimes of PC) is bound to arachidonic acid. [Note: Barth syndrome, a rare X-linked disorder characterized by cardiomyopathy, muscle weakness, and neutropenia, is the result of defects in cardiolipin remodeling.]
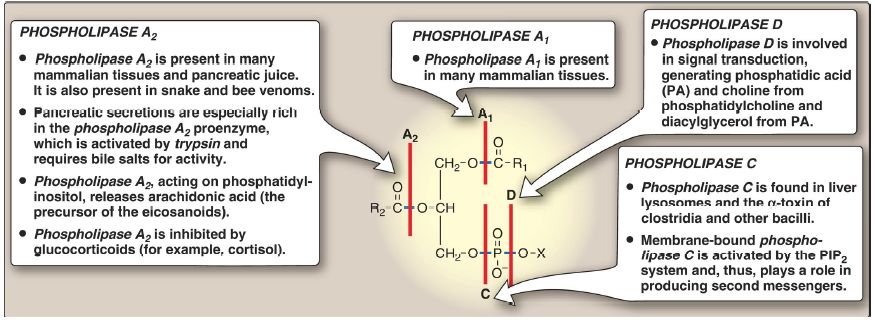
Figure 1: Degradation of glycerophospholipids by phospholipases. PIP2 = phosphatidylinositol 4,5-bisphosphate; R1 and R2 = fatty acids; X = an alcohol.
B. Sphingomyelin
Sphingomyelin is degraded by sphingomyelinase, a lysosomal enzyme that removes phosphorylcholine, leaving a ceramide. The ceramide is, in turn, cleaved by ceramidase into sphingosine and a free FA (Fig. 2). [Note: The released ceramide and sphingosine regulate signal transduction pathways, in part by influencing the activity of protein kinase C and, thus, the phosphorylation of its protein substrates. They also promote apoptosis.] Niemann-Pick disease (types A and B) is an autosomal-recessive disorder caused by the inability to degrade sphingomyelin due to a deficiency of sphingomyelinase, a type of phospholipase C. In the severe infantile form (type A, which shows <1% of normal enzymic activity), the liver and spleen are the primary sites of lipid deposits and are, therefore, greatly enlarged. The lipid consists primarily of the sphingomyelin that cannot be degraded (Fig. 3). Infants with this lysosomal storage disease experience rapid and progressive neurodegeneration as a result of deposition of sphingomyelin in the CNS, and they die in early childhood. A less severe variant (type B, which shows up to 10% of normal activity) with a later age of onset and a longer survival time causes little to no damage to neural tissue, but lungs, spleen, liver, and bone marrow are affected, resulting in a chronic form of the disease. Although Niemann-Pick disease occurs in all ethnic groups, type A occurs with greater frequency in the Ashkenazi Jewish population.
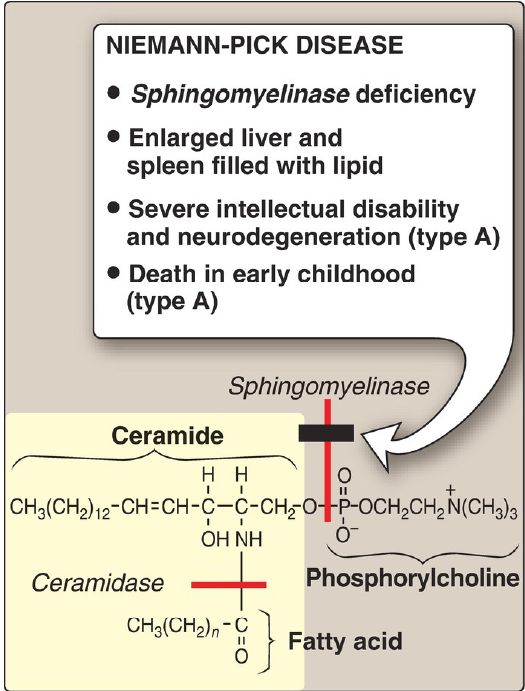
Figure 2: Degradation of sphingomyelin. [Note: Type B is the nonneuropathic form. It has a later age of onset and a longer survival time than type A.]
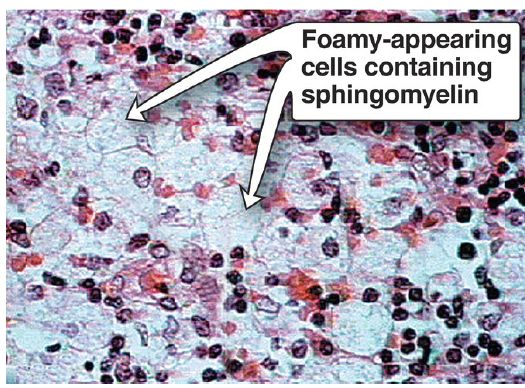
Figure 3: Accumulation of lipids in spleen cells from a patient with Niemann-Pick disease.

|
|
|
|
"عادة ليلية" قد تكون المفتاح للوقاية من الخرف
|
|
|
|
|
|
|
ممتص الصدمات: طريقة عمله وأهميته وأبرز علامات تلفه
|
|
|
|
|
|
|
المجمع العلمي للقرآن الكريم يقيم جلسة حوارية لطلبة جامعة الكوفة
|
|
|