The Stepwise Ionic Mechanism, Halogen Addition
We shall give particular attention here to the addition of bromine to alkenes because this reaction is carried out very conveniently in the laboratory and illustrates a number of important points about electrophilic addition reactions. Much of what follows applies to addition of the other halogens, except fluorine.
A significant observation concerning bromine addition is that it and many of the other reactions listed above proceed in the dark and are not influenced by radical inhibitors. This is evidence against a radical-chain mechanism of the type involved in the halogenation of alkanes. However, it does not preclude the operation of radical-addition reactions under other conditions, and, as we shall see later in this chapter, bromine, chlorine, and many other reagents that commonly add to alkenes by ionic mechanisms also can add by radical mechanisms.
One alternative to a radical-chain reaction for bromine addition to an alkene would be the simple four-center, one-step process shown in Figure 10-7.

Figure 10-7: Representation of a one-step suprafacial mechanism for addition of bromine to ethene. Gas-phase additions appear to proceed in this manner.
The mechanism of Figure 10-7 cannot be correct for bromine addition to alkenes in solution for two important reasons. First, notice that this mechanism requires that the two C−Br bonds be formed on the same side of the double bond, and hence produce suprafacial addition. However, there is much evidence to show that bromine and many other reagents add to alkenes to form antarafacial addition products (Figure 10-8).

Figure 10-8: Antarafacial addition of bromine to ethene, usually observed in solution.
Cyclohexene adds bromine to give trans-1,2-dibromocyclohexane:
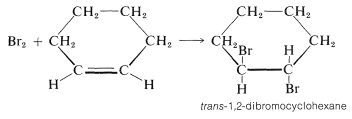
The cis isomer is not formed at all. To give the trans isomer, the two new C−Br bonds have to be formed on opposite sides of the double bond by antarafacial addition. But this is impossible by a one-step mechanism because the Br−Br bond would have to stretch too far to permit the formation of both C−Br bonds at the same time.
The second piece of evidence against the mechanism of Figure 10-7 is that bromine addition reactions carried out in the presence of more than one nucleophilic reagent usually give mixtures of products. Thus the addition of bromine to an alkene in methanol solution containing lithium chloride leads not only to the expected dibromoalkane, but also to products resulting from attack by chloride ions and by the solvent:
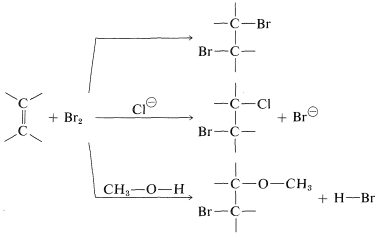
The intervention of extraneous nucleophiles suggests a stepwise mechanism in which the nucleophiles compete for a reactive intermediate formed in one of the steps.
A somewhat oversimplified two-step mechanism that accounts for most of the foregoing facts is illustrated for the addition of bromine to ethene. [In the formation shown below, the curved arrows are not considered to have real mechanistic significance, but are used primarily to show which atoms can be regarded as nucleophilic (donate electrons) and which as electrophilic (accept electrons). The arrowheads always should be drawn to point to the atoms that are formulated as accepting a pair of electrons.]
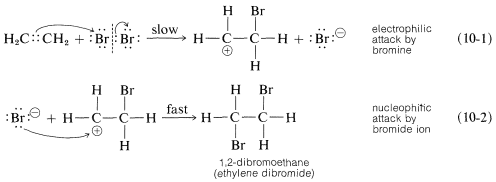
The first step (which involves electrophilic attack by bromine on the double bond) produces a bromide ion and a carbocation, as shown in Equation 10-1.
As we know from our study of SN1 reactions , carbocations react readily with nucleophilic reagents. Therefore in the second step of the bromine-addition mechanism, shown in Equation 10-2, the bromoethyl cation is expected to combine rapidly with bromide ion to give the dibromo compound. However, if other nucleophiles, such as Cl⊖ or CH3OH, are present in solution, they should be able to compete with bromide ion for the cation, as in Equations 10-3 and 10-4, and mixtures of products will result:
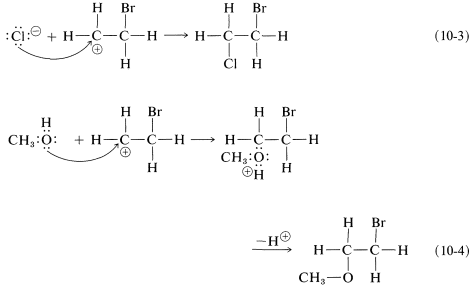
To account for the observation that all of these reactions result in antarafacial addition, we must conclude that the first and second steps take place from opposite sides of the double bond.





















