آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Contact Maps |


|
|
Read More
Date: 24-4-2021
Date: 13-12-2015
Date: 12-6-2021
|
Contact Maps
Contact maps are two-dimensional representations of three-dimensional protein structures. A three-dimensional description of a protein structure composed of N structural units could be expressed as an N × N array of the pairwise distances. This could be done for all pairs of atoms, for selected types of atoms (eg, Ca atoms), for groups of atoms (eg, side-chain centers of mass), or for entire amino-acid residues. Contact maps are generated from such matrices by taking a certain cutoff value for the pairwise distances. For example, the N × N matrix of the distances between protein Ca atoms (1, 2) can be transformed into a Ca-based contact map (3). Those Ca atoms that are closer to each other in the protein structure than the chosen cutoff distance are considered to be “in contact.” This produces a binary N × N matrix—a so-called black and white contact map. Alternatively, one may assume a set of several critical values for the distances between Ca atoms (or for other atoms or groups of atoms) and generate an integer matrix—the equivalent of a contact map with a colored or gray scale. A map of main-chain hydrogen bonds could also be considered as a variant of a protein contact map. The choice of structural units being mapped and the choice of cutoff distances determine the quality and range of structural information being stored in a contact map (see Fig. 1) .

Figure 1. Schematic drawing of the structure of the B domain of protein G and its contact maps. (a) Three-dimensional structure of the B domain. (b) Ca-based contact maps: above the diagonal, the black and white map with a cutoff distance of 8 Å; below the diagonal is the gray-scale map where various shades of gray correspond to three values of the cutoff distance, 8 (darkest), 10, and 12 Å. (c) Side-chain-based contact map. Above the diagonal, the dark squares correspond to the pairs of side chains for which the distance between at least one pair of heavy atoms is less than 5 Å. Below the diagonal, a 6.25-Å cutoff criterion has been applied to the centers of mass of the side chains. (d) The main chain hydrogen bond map for the same structure. The amino acid sequence of the protein is given along each axis in one-letter code, and the secondary structure is indicated as follows: E is extended b-strand, H is helix, and S and T are two types of turns.
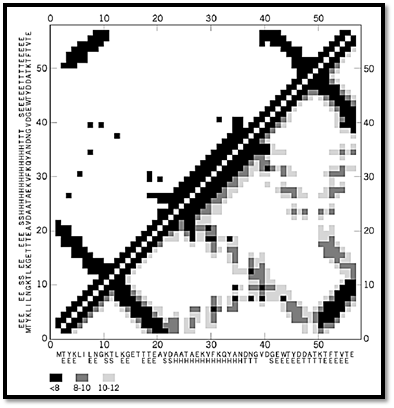
Figure 2. (continued) Schematic drawing of the structure of the B domain of protein G and its contact maps. (a) Three-dimensional structure of the B domain. (b) Ca-based contact maps: above the diagonal, the black and white map with a cutoff distance of 8 Å; below the diagonal is the gray-scale map where various shades of gray correspond to three values of the cutoff distance, 8 (darkest), 10, and 12 Å. (c) Side-chain-based contact map. Above the diagonal, the dark squares correspond to the pairs of side chains for which the distance between at least one pair of heavy atoms is less than 5 Å. Below the diagonal, a 6.25-Å cutoff criterion has been applied to the centers of mass of the side chains. (d) The main chain hydrogen bond map for the same structure. The amino acid sequence of the protein is given along each axis in one-letter code, and the secondary structure is indicated as follows: E is extended b-strand, H is helix, and S and T are two types of turns.
1. Contact Maps as a Fingerprint of Protein Three-Dimensional Structure
A contact map constitutes a structural “fingerprint” of a protein (4). Each protein can be identified based on its contact map. The secondary structure, fold topology, and side-chain packing patterns (for side-chain contact maps) can be visualized conveniently and read from the contact map. Furthermore, structural similarity between a pair of proteins is immediately apparent by a very pronounced similarity of their contact maps; in comparing two protein structures, there is no need to search all their possible relative orientations. The reconstruction of a protein structure from its contact map is more complex, although low-to-moderate resolution three-dimensional models can be easily built, even from a fragmentary contact map (5). The accuracy of the model depends on the type of contact map and the computational tools employed. A combination of protein nuclear magnetic resonance NMR spectra constitutes a hybrid contact map of a protein, and model building from these data is an example of a map-to-structure modeling procedure (6, 7).
2. Ca-Based Contact Maps
Ca-based contact maps and distance matrices were perhaps the first commonly used maps for visualization of protein structures (3, 8, 9). An example is given in Figure 1b. These contact maps reflect well the overall topology of the protein fold, but only rather coarse structural details can be read from them. This is due to the fact that the Ca–Ca distance distributions extracted from protein structures have several convoluted peaks. These peaks correspond to various distances between pairs of various secondary structure elements (a-helices, beta-strands, etc.). Thus, a single cutoff distance is always inadequate: Too small a value would miss some helix-to-helix contacts, while too large a value may create some problems with identification of the secondary structure patterns. Gray scale maps (several cutoff ranges) communicate much more detailed structural information.
3. Side-Chain Contact Maps
Side-chain contact maps contain much richer information, not only about the topology of a protein fold and its secondary structure, but also many fine details about the packing patterns of the protein side-chains. Various conventions can be used to build side-chain contact maps; two are illustrated in Figure 1. In one case, two residues are assumed to be in contact when any two heavy atoms (ie, all except hydrogen) are a shorter distance from each other than some assumed cutoff. Due to the comparable size of all the united atom types constituting the side chains (eg, CH2, CH, NH2, etc.), a good choice of cutoff distance is between 4.5 and 5.0 Å (4, 10). In this range, the number of detected contacts is not sensitive to the particular choice of cutoff, and the packing pattern of the side chains is always described with high fidelity. Characteristic patterns of contacts between elements of secondary structure are an important and useful feature of these contact maps (11). Alternatively, one may build a side-chain contact map using the side-chain centers of mass as a reference. In this case, a larger value of the cutoff distance needs to be used. These cutoff values for the distance between side-chain centers of mass could be made specific for certain amino acid pairs, on the basis of the different sizes of the side chains, which produce different average contact distances for various pairs. As seen in Figure 1, the two approaches (atom based and center of mass based) lead to very similar protein representations. The patterns of the atom-based contact maps are slightly better defined.
4. Regularities of the Contact Maps Reflect Regularities of Protein Structures Different types of contact maps reflect different aspects of the regularities seen in protein structures. The side-chain-based contact maps are a very good example (10). Near the diagonal of the map, the distinct features of the protein secondary structure can be easily read. Indeed, for extended fragments of the polypeptide chain, only residues i and i + 2 can be in contact. For a-helices, the i, i + 3 and i, i + 4 patterns of contacts are well pronounced. Furthermore, characteristic clusters of contacts further away from the diagonal reflect the packing between particular pairs of b-strands within the b-sheets. Parallel and antiparallel structures have very different features on the contact patterns. Very characteristic patterns could also be observed for other pairs of secondary-structure elements. It is even very easy to distinguish between the patterns for two helices in a helical or a/b protein and in the coiled-coil structural motifs. Figure 2 shows some typical side-chain contact map patterns.

Figure 3. Representative patterns of side-chain contact maps describing interactions between a-helices (top) and between b-strands in parallel and antiparallel beta-sheets (bottom).
5. Application of Contact Maps in Modeling Protein Structure
Contact maps provide a very convenient way of identifying pairwise interactions within protein structures. This has been applied in various algorithms for threading protein sequences, where contact maps are actually used as ersatz template structures (4). In computer simulations of biological molecules, contact maps provide a convenient way of displaying structural changes. Also, regularities of the patterns seen in all classes of proteins can provide a guideline for designing knowledge-based multibody potentials (12, 13) and for protein modeling in a reduced (and perhaps also in all-atom) representation (13, 14). Such potentials may be necessary to reproduce the all-or-none character of protein folding transitions in model simulations (15).
One can easily recognize the distinct features of well-defined contact maps, with their characteristic patterns, after inspecting several maps of various proteins. This is an excellent example of a pattern recognition problem that could be learned by neural network calculations. Then such a trained network can be used for the automated recognition of good versus poor models of protein structure (16).
References
1. I. D. Kuntz (1975) J. Am. Chem. Soc. 97, 4362–4366.
2. F. M. Richards and C. E. Kundrot (1988) Proteins 3, 71–84.
3. M. Levitt (1976) J. Mol. Biol. 104, 59–107.
4. A. Godzik, J. Skolnick, and A. Kolinski (1992) J. Mol. Biol. 227, 227–238.
5. J. Skolnick, A. Kolinski, and A. R. Ortiz (1997) J. Mol. Biol. 265, 217–241.
6. W. Braun and N. Go (1985) J. Mol. Biol. 186, 611–626.
7. R. Kaptein, R. Boelens, R. M. Scheek, and W. F. van Gunsteren (1988) Biochemistry 27, 5389–5395.
8. M. N. Liebman, C. A. Venanzi, and H. Weinstein (1985) Biopolymers 24, 1722–1758.
9. D. C. Philips (1970) Biochem. Soc. Symp. 30, 11–28.
10. A. Godzik and C. Sander (1989) Protein Eng. 2, 589–596.
11. A. Godzik, J. Skolnick, and A. Kolinski (1993) Protein Eng. 6, 801–810.
12. A. Kolinski, A. Godzik, and J. Skolnick (1993) J. Chem. Phys. 98, 7420–7433.
13. A. Kolinski and J. Skolnick (1996) Lattice Models of Protein Folding, Dynamics and Thermodynamics, R. G. Landes, Austin, TX.
14. K. A. Olszewski, A. Kolinski, and J. Skolnick (1996) Proteins 25, 286–299.
15. A. Kolinski, W. Galazka, and J. Skolnick (1996) Proteins 26, 271–287.
16. M. Milik, A. Kolinski, and J. Skolnick (1995) Protein Eng. 8, 225–236.

|
|
|
|
دخلت غرفة فنسيت ماذا تريد من داخلها.. خبير يفسر الحالة
|
|
|
|
|
|
|
ثورة طبية.. ابتكار أصغر جهاز لتنظيم ضربات القلب في العالم
|
|
|
|
|
|
|
بالصور: ممثل المرجعية العليا الشيخ عبد المهدي الكربلائي يؤم جموع المؤمنين في صلاة عيد الفطر المبارك داخل الصحن الحسيني الشريف
|
|
|