آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Complement System |


|
|
Read More
Date: 5-6-2021
Date: 29-5-2021
Date: 11-12-2020
|
Complement System
1. Introduction
After the discovery of humoral immunity, Hans Buchner found that if fresh serum possessing an antibacterial antibody was added to bacteria, the bacteria were quickly lysed. However, if the serum was heated to 56°C, the lytic capacity of the serum was lost. He also demonstrated that the loss of lytic capacity could be restored by using unheated nonimmune serum in conjunction with heated immune serum. These studies were extended by others, including Jules Bordet, who concluded that serum contained two components necessary for cellular lysis. These two components are heat-stable antibodies and a heat-labile component that “complements” the lytic function of antibodies. He reasoned that antibodies had two binding sites, one for antigen and the other for heat-labile substance that was given the name complement.
Complement is now known not to be a single component, but it consists of at least 20 chemically and immunologically distinct plasma proteins capable of interacting with one another in a highly regulated manner to provide at least four main biological functions. First, cytolysis is mediated by the association, or polymerization, of specifically activated complement components on the surface of targets cells. These components form pores that disrupt the integrity of the lipid membrane, and the cell is killed by osmotic lysis. Second, antibody and antigen combine to form immune complexes that, unless removed, can result in damage of body tissues. The binding of complement proteins prevent the damage from immune complexes by mediating their solubilization and clearance. Third, the opsinization of foreign particles is mediated by the binding of complement proteins (opsonins.( Phagocytic leukocytes bear receptors for these complement proteins, so that opsinized particles are cleared from the body by phagocytosis. Finally, through the activation of complement, proteolytic fragments are released whose function are to mediate inflammation. These fragments, or “anaphylatoxins,” can act on several target cells such as neutrophils, smooth muscle, and vascular endothelium, as well as organ systems of the body.
The complement proteins are normally present in the circulation and are produced primarily by the liver and other extrahepatic sources, such as macrophages and fibroblasts. Some of the components are produced as functionally inactive proteinases that are activated only when proteolytically cleaved themselves by previously activated complement proteins. Complement activation occurs only at localized sites under specific conditions. The binding of specific antibody to antigen can initiate complement activation through what is called the classical pathway. Additionally, in the absence of antibody, some complement components are directly activated by binding to the surfaces of infectious agents, such as bacteria, fungi, and viruses, through what is called the alternative pathway.
2. Overview of the Complement System
Individual proteins of the complement system are present in the serum as functionally inactive molecules. The individual precursor proteins are designated numerically, such as C1, C2, up to C9. Other proteins involved with the complement system have retained their common, or trivial, names such as properdin, factor B, and factor H. Individual components must be activated sequentially and specifically for the complement cascade to progress and mediate inflammation and lysis of foreign organisms. The activation of complement is a dynamic process that allows the proteins of the system to interact functionally and is not a static singular event.
An overview of the operation of the complement system is shown in Figure 1, and biochemical and physical information of the proteins involved in the complement system are listed in Table 1. A protein called C3 is the central component of the complement system. The alternative and classical pathways are two parallel, but entirely independent, mechanisms that lead to the formation of C3 convertases whose function is to cleave C3 into C3a and C3b. In the classical pathway, initial events involve binding of specific antibody to antigen. This antigen–antibody complex binds complement C1 and sequentially activates complement proteins C4 and C2, leading to the formation of the C4b2b complex that functions as the classical pathway C3 convertase. In contrast, activation of complement via the alternative pathway occurs independent of antigen–antibody complexes. Instead, alternative pathway activation is dependent on activators that allow the formation and deposition of the C3bBb complex (alternative pathway C3 convertase) on their surfaces. Activators of the alternative pathway include many bacteria, fungi, and viruses. Both classical and alternative convertases can cleave additional C3, generating more C3b (see Fig. 1). The binding of additional C3b to these C3 convertases changes them conformationally to C5 convertases, which specifically cleave C5. Once cleaved, both the alternative and classical pathways share the same terminal steps. These steps do not involve the proteolytic cleavage of additional components but instead involve the sequential binding of components C6, C7, C8, and C9. This terminal assembly leads to the formation of the membrane attack complex (MAC), resulting in the osmotic lysis or cytolysis of bacteria or other affected cells.
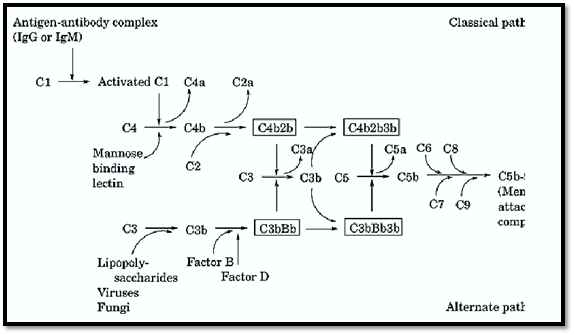
Figure 1. Schematic representation of the alternative and classical activation of the complement pathway leading to the assembly of the membrane attack complex.
Table 1. Protein Components of the Classical and Alternative Pathwaysa
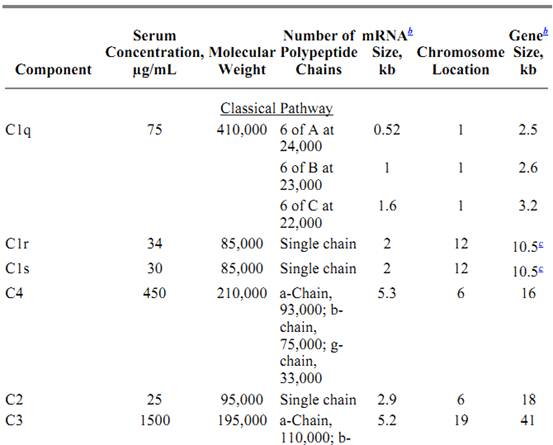
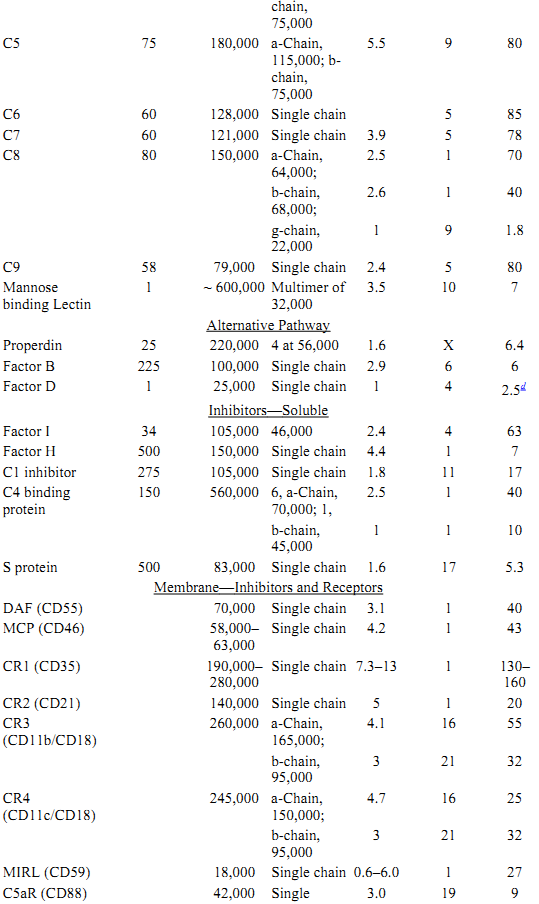
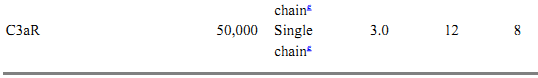
a All data for proteins listed are for human complement components.
b Approximate size.
c C1r size estimated from proximity and homology to C1s.
d Complete factor D cDNA hybridizes within a 2.5-kb genomic fragment.
e Single chain with 7-transmembrane domains.
During activation, through either the classical or alternative pathway, various cleavage products mediate a variety of biological functions. As discussed, cytolysis is mediated through the membrane attack complex. Opsinization and subsequent phagocytosis by leukocytes is mediated primarily through C3b, a cleavage fragment of C3 that binds receptors for C3b. Cleavage products of C3, C4, and C5 (C3a, C4a, and C5a anaphylatoxins) mediate inflammatory events and the recruitment and activation of leukocytes.
3. Assembly and Activation of the Classical Pathway
The classical pathway is activated by antigen–antibody complexes or aggregated immunoglobulin acting on complement component C1 (Table 1). C1 consists of three distinct protein molecules, C1q, C1r, and C1s, which are held together by Ca2+-dependent bonds. C1 is present in serum as a firm C1q-C1r-C1s complex, while individual C1 components are found only in pathologic conditions. C1 contains one molecule of C1q and two molecules of both C1r and C1s.
A molecule of C1q is comprised of 18 polypeptide chains of three distinct types (A, B, and C) which are twisted together comprising one subunit. Each C1q molecule is then made up of such six subunits connected together; once assembled, its structure is similar to that of collagen. The C1q molecule bears the sites that enable the entire C1 molecule to bind the Fc region of IgM and IgG immunoglobulin molecules. A single C1 molecule can bind about six IgG molecules. After binding to Ag-Ab complexes, C1q undergoes a conformational change that causes C1r to activate itself by a limited self-cleavage.
Activated C1r cleaves a single peptide bond in C1s, which then acquires enzymatic activity of its own. This newly generated enzyme is a serine esterase type and mediates the cleavage of component C4. With the activation of the C1s enzyme, the initial process is complete, and the earlier reactants, including antibody, antigen, C1q, and C1r, are no longer needed.
C4 is composed of three nonidentical polypeptide chains and is synthesized as a single-chain precursor in a beta–alpha–gamma orientation. C1 cleaves a single bond on the C4 alpha chain leading to the production of C4a and C4b. This cleavage leads to the formation of a labile binding site in the larger fragment of C4b, which enables it to bind to the activating surface (activator). The C4b alpha chain, like C3, contains an internal thioester bond formed between a glutamic acid and cysteine residue. Cleavage of the alpha chain of C4 is followed by stress-induced hydrolysis of the thioester bond. This permits the reactive acyl group of the glutamyl residue to form a covalent bond with a reactive hydroxyl or amino group on the surface of the activator.
C2 cleavage by C1s also generates a labile binding site of unknown chemical composition in the larger C2b fragment, which allows it to bind to C4b. Mg2+ ions are required for the formation of the C4b2b complex. Formation of the C4b2b complex is not very efficient, as the majority of C2 and C4 molecules entering into this reaction lose their labile binding sites before achieving union with membranes or with each other and diffuse away as inactive reaction products. The newly formed C4b2b is a proteolytic enzyme that assumes the role of continuing the complement reaction cascade, so earlier activating components are no longer required. The C4b2b complex is also called the classical C3 convertase, which acts to cleave C3.
The substrate for C4b2b is C3, which is synthesized as a single chain of beta–alpha orientation that is processed after translation. The larger, alpha chain, is cleaved at a single site located near the amino terminus. The smaller resulting fragment C3a (9000 Mr (relative molecular mass) is a biologically potent peptide that mediates inflammation and will be discussed later. A labile binding site is generated in the larger fragment C3b, which enables the molecule to attach to membranes at sites near, but distinct from, those utilized by antibody and C4b2b. Often described as the “lynchpin,” C3 is the precursor of several biologically active fragments that function by association with the other proteins of the complement system leading to lysis of the target cell cell via either classical or alternative pathways. C3 has numerous distinct binding sites, one of which is the thioester domain that allows the molecule to bind covalently to target sites and particles, such as immune complexes and membrane surfaces.
The chemical site of the C3 thioester has the sequence Gly-Cys-Gly-Glu-Glu-Asn with the Cys and second Glu residues joined by a thioester bond, specifically a b-cysteinyl–g–glutaminyl thioester bond. This thioester bond is present in the C3d domain of the alpha chain. With the cleavage of C3 into C3a and C3b, the thioester undergoes a stress-mediated hyrdolysis, and the reactive acyl group of the glutamyl residue forms a covalent bond with a reactive hydroxyl or amino group on the activator surface. A major amount of reactive C3 fails to achieve binding with activators as most of these reactive thioesters have reacted with water.
The attachment of C3b to membranes in the vicinity of C4b2b molecules leads to the generation of the last enzyme of the classical pathway, C4b2b3b, or the classical C5 convertase. To this end, the classical pathway has finished the initial steps of activation. From here, the C5 convertase acts on C5 to initiate steps in the forming of the MAC.
4. Activation of the Classical Pathway
The activation of the classical pathway can occur by one of two ways: activation through the use of immunoglobulin or through nonimmunoglobulin activation by cleavage of C1. Immunoglobulin activation of the classical pathway involves the use of antibodies belonging to subclasses IgG1, IgG2, and IgG3, as well as IgM, are capable of initiating the classical pathway. Immunoglobulins IgG4, IgA, IgD, and IgE are inactive in this regard. Among the three listed above, IgG3 is the most active in interacting with C1, then IgG1 and IgG2. Activation occurs when the first component C1 binds to a site in the Fc region of the IgG or IgM. Only one molecule of bound IgM is capable of initiating the activation of the classical pathway; however, it is estimated that six molecules of IgG are required. Nonimmunologic activation of the classical pathway can be accomplished by diverse substances such as DNA, certain viruses, and trypsin-like enzymes, by direct proteolytic attack on the C1 molecule. Of particular interest are lectins that also act as a nonimmunological activator of the classical pathway. Mannose binding lectin (MBL) is a serum protein found in all mammals and is regulated as an acute-phase protein. MBL is capable of binding carbohydrate moities present on many microorganisms and subsequently capable of activating the classical pathway through C4 without the need for specific antibody or C1q.
5. Alternative Complement Pathway or Properdin Pathway
The alternative pathway may be activated immunologically by aggregates of human IgA and by certain complex polysaccharides, fungi, viruses, bacterial lipopolysaccharides, and trypsin-like enzymes. This system was originally described as the properdin system, a group of proteins involved in resistance to infection, but distinct from complement. The proteins of the alternative pathway, C3, factor B, factor D, and properdin (factor P) perform the functions of activation, recognition, and amplification of the pathway, resulting in the formation of the activator bound C3/C5 convertase. The alternate pathway system was found to be involved in the destruction of certain bacteria, neutralization of some viruses, and red blood cell of (RBC) lysis from patients with paroxysmal nocturnal hemoglobinuria.
6. Activation of the Alternate Pathway
Alternate pathway activation initially proceeds in a manner different from that for the classical pathway. The exact mechanism by which the first C3b molecule is produced is still controversial. However, it is clear that antibody is not required since mixtures of only highly purified alternative pathway proteins behave as well as serum. The ultimate aim of the proteins, C3, factor B, factor D, and properdin is the initiation, recognition, and amplification of the pathway that results in the formation of the activator bound C3/C5 convertases. The first requirement for activation is the presence of C3, which participates in the initiation and amplification of the pathway. It is generally accepted that activated C3 (but not-yet cleaved) [C3*] reacts with proenzyme B, and this complex is then cleaved by factor D, yielding two fragments, Ba and Bb. Bb then attaches to C3b forming the alternative pathway C3 convertase. The C3 convertase is able to cleave additional C3 into C3a and C3b. While most C3b remains in the fluid phase, some binds to various cell surfaces. Thus C3b is continuously generated and is deposited on the surface of the activator. The alternative C3 convertase is unstable and decays rapidly unless another alternative pathway member, properdin, binds to the C3 convertase and stabilizes it. Properdin binding of C3bBb extends its functional half-life eightfold. The function of the C3 convertase is to produce enough meta-stable C3 to deposit on the surface of surrounding particles. Surface-bound C3b then interacts with factor B and is activated by D to form more C3bBb. This enzyme is capable of cleaving large amounts of C3, and forms more C3bBb (C3bBb3b); thus, a positive feedback mechanism is formed that amplifies the initial stimulus. Many of the C3b molecules bind to the surface of the activator in close proximity to these enzymes, forming C3bBb3b or the alternative pathway C5 convertase. Like its counterpart in the classical pathway (C4b2b3b), the function of C3bBb3b is to proteolytically cleave C5 and initiate the assembly of the membrane attack complex.
7. The Membrane Attack Complex: C5–C9 Reaction
The terminal portion of the complement sequence is known as the MAC (complex). C5–C9 must become membrane bound for damage to occur. This complex may attach to a cell bearing the activation enzymes of the classic or alternate pathways, or it may attach to a bystander cell. The MAC is initiated by the cleavage of C5 by the alternative and classical C5 convertases.
Similar to C3, pro-C5 is secreted in a beta–alpha orientation prior to post translational modification. C5 shares sequence homology with C3 and C4, including the domain corresponding to the thioester region; however, C5 lacks the essential cysteine and glutamic acid residues necessary for thioester formation. Assembly of the MAC initiates with the cleavage of C5. Cleavage at residue 74 of the alpha chain, results in the generation of C5a anaphylatoxin and C5b (Table 1), where C5b can then bind C6. C6 is a single chain peptide that, when bound to C5b, remains loosely attached to the membrane until the addition of C7. Once a single C7 has bound the C5b6, the resulting complex is highly lipophilic and inserts into the lipid bilayer. When inserted, the C5b67 complex serves as a high affinity integral membrane receptor for a single C8 molecule. C8 is composed of three nonidentical polypeptide chains and has an interesting structure in that the alpha and gamma chains are covalently linked by one or more disulfide bonds and the beta chain is joined to the alpha–gamma chains by noncovalent forces. The C5b-8 becomes stably attached to the membrane by insertion of the C8 gamma chain into the lipid bilayer. With this early “MAC” formed, cellular leakage can occur at this stage. The cytolytic process is greatly accelerated by the attachment of C9, the final complement component. The C9 monomer polymerizes at the site of the C5b-8 complex. As few as 4 C9 molecules can result in lysis for many microorganisms. However, when the MAC contains as many 12 to 15 C9 molecules, the “poly-C9” forms a pore in the membrane and permits passive exchange of cytoplasmic contents with the extracellular media with subsequent osmotic lysis. The pores have an internal diameter of about 110 Å and appear similar in structure to those formed by lytic T cells when the related protein perforin is deposited on target cells.
8. Control Mechanisms of the Complement System
Given the lytic potential of the complement system, uncontrolled activation can lead to the formation of the MAC on normal tissues and significant generation of C3a, C4a, and C5a anaphylatoxins. Multiple fluid phase and membrane bound proteins tightly regulate both the alternative and classical pathways by acting at specific steps during activation. In addition, the uncontrolled activation of the complement cascade is also prevented by the necessity of specific activators, such as antibody–antigen complexes, as well as the lability of the activated complement components such as C3b. Deficiencies in regulatory proteins result in unregulated complement activation and are apparent in various disease conditions. An overview of the regulation complement pathway activation by both membrane-bound and soluble inhibitors are shown in Figure 2. The biochemical and physical information of these inhibitors are listed in Table 1.
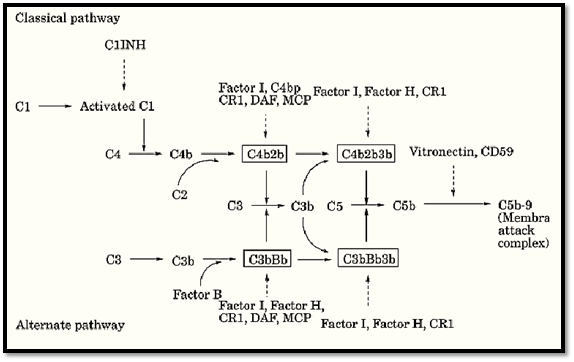
Figure 2. Regulation of classical and alternative complement pathway activation by membrane and soluble inhibitors. Principal components and C3/C5 convertases are retained from Figure 1. Regulators of complement activation are shownitalics.
9. Classical Pathway Inactivators
A protein celled C1 inhibitor (C1INH) acts to inhibit the initiation of the classical pathway. C1INH is a member of the serine protease inhibitor family and acts by blocking C1r and C1s of the C1 complex. As with other serpins, such as a1-antitrypsin, C1INH uses a “bait” sequence that mimics the normal substrates of C1r and C1s. However, when C1INH is cleaved by C1r or C1s, it forms a covalent ester linkage that inhibits cleavage of C4 and C2, thus inhibiting the progression of the classical pathway. Most of the C1 in the serum is already bound to C1INH. On specific binding of C1 to antigen–antibody complexes, C1INH is released allowing classical pathway activation to proceed.
Other means to control the classical pathway involve interfering with the formation of the C3 convertase. Such inhibition can be mediated either through interfering with the assembly of the convertase or in promoting the dissociation of the convertase. Three proteins called C4-binding protein (C4 bp), complement receptor type I (CR1) (also known as CD35), and decay accelerating factor (DAF) (also known as CD55) function as inhibitors of the classical pathway by binding to C4b. In doing so, they compete with C2 for C4b and thus inhibit the formation of the classical C3 convertase. In binding C4b, they also promote the dissociation of the C3 convertase. All three proteins are structurally similar and are homologous members of a family of proteins called the regulators of complement activity (RCAs.(
Another way that the C3 convertase can be inhibited is by the cleavage of C4b by a protein called factor I. Factor I is a serine esterase that cleaves C4b into two additional fragments: C4c and C4d. C4d remains bound to the original activating surface; however, it is unable to contribute to the formation of the classical C3 convertase. Cleavage of C4b by factor I can only occur in the presence of a “cofactor.” The proteins that can serve a cofactor for factor I cleavage include CR1, C4b, and membrane cofactor protein (MCP), or CD46. MCP is also a member of the RCA family but does not possess decay accelerating activity.
10. Alternative Pathway Inactivators
Regulation of the alternative pathway is also accomplished by several circulating and membrane bound proteins, many of which also act on the classical pathway. In analogous fashion, the inhibition of convertase activity through either decay acceleration or by providing cofactor activity for factor I are the major ways in which this is accomplished.
Factor H is a soluble serum protein that possesses decay-accelerating activity that is specific for the Bb fragment of the alternative pathway. By competitively binding Bb, the formation of the alternative C3 covertase is impaired. Factor H is also a member of the RCA family and in binding Bb can also promote the dissassembly of the alternative C3 convertase.
In addition to its role in inhibiting the classical pathway activation, factor I can also cleave C3b using factor H, MCP, and CR1 as cofactors. The resulting fragment of C3b (iC3b) is unable to participate in the formation of the alternative C3 convertase.
11. Inactivators of the Membrane Attack Complex
With the alternative and classical pathways activated, the potential for normal or bystander host cells to be subjected to lysis is significant. Excessive cell lysis is normally prevented by a number of proteins that act to inhibit the formation of the membrane attack complex.
A membrane bound protein that is capable of inactivating the membrane attack complex is the membrane inhibitor of reactive lysis (MIRL), more commonly known as CD59. CD59 is broadly expressed among different cell types and is probably the most important molecule in minimizing damage to bystander cells. It is thought that CD59 acts by binding C8 and C9 and thus inhibits the polymerization of C9 as well as the insertion of the MAC into the membrane.
As discussed above, when C7 binds C5b6, the resulting complex is lipophilic and it inserts into the membrane. This process of membrane insertion is inhibited by S protein, also known as vitronectin, a protein related to fibronectin and laminin. The S protein is thought to function by binding the C5b67 complex before it inserts into the membrane.
12.Complement Receptors and Biological Consequences of Complement Activation
Through the activation of the complement cascade, through either the classical or alternative pathways, numerous complement derived fragments are generated. Many of these fragments possess biological activities that are mediated through the use of specific receptors.
In addition to regulating the activation of complement, CR1 (CD35) serves as a high affinity receptor for C3b and C4b. CR1 functions as a receptor for C3b- and C4b-coated particles wherein macrophages ingest and remove these coated particles. Another receptor, CR2 (CD21), is expressed primarily on B cells, dendritic cells, and epithelial cells. CR2 is known to specifically bind iC3b and C3dg—both factor I–generated cleavage products. In addition, CR2 is also known as the Epstein–Barr virus receptor. Mac-1 or CR3 (CD11b/CD18) is related to the integrin family and is a specific receptor for the factor I cleavage product of C3b, iC3b. CR3 is expressed on many different cells derived from the bone marrow including neutrophils, mononuclear phagocytes, mast cells and natural killer (NK) cells. The primary role of CR3 is the phagocytosis and clearance of iC3b-coated particles. CR4 (CD11c/CD18), like CR3, is also a member of the integrin family and also binds iC3b as well as C3dg.
The cleavage of C3, C4, and C5 results in the production of C3a, C4a, and C5a anaphylatoxins. These hormone-like peptides mediate a variety of cellular and biochemical responses from leukocytes, including the generation of bacteriocidal superoxide radicals, proteolytic enzyme release from intracellular granules, cellular aggregation, smooth muscle contraction, and phagocytosis. In addition, C5a induces chemotaxis, or the migration, of leukocytes into areas of complement activation. The induction of chemotaxis was thought to be unique for C5a; however, recent findings suggest that C3a may be chemotactic for eosinophils, a cell whose function is mediating allergic reactions. These anaphylatoxins mediate their effects through specific receptors that are coupled to GTP-binding proteins and whose signal transduction can be abrogated by pertussis toxin. The C3a and C4a receptors have been found to be expressed on mast cells, basophils, lymphocytes, and smooth muscle cells. The C5a receptor has been found on mast cells, basophils, neutrophils, monocytes/macrophages, endothelial cells, smooth muscle, liver parenchyma, and epithelial lining of lung as well as astrocytes and microglia of the central nervous system. Of the three anaphylatoxins, the most potent is C5a in mediating biological effects. C3a is approximately 20-fold less potent, while C4a is about 2500-fold less potent than C5a. C5a has been shown to stimulate the release of tumor necrosis factor (TNF) from mast cells as well as stimulating P-selectin (CD62P) expression on vascular endothelium to promote neutrophil binding.
13. Complement Genes and Relationships
On the basis of sequence homologies, many of the complement proteins can be grouped together as members of families. Members of these families share structural and/or functional similarities by which relationships can be assessed.
One of the more defined families is that of the regulators of complement activity (RCAs) as previously discussed. Factor H, CR1, CR2, DAF, C4 bp, and MCP are all members of the RCA, and all share the ability to bind both C3b and C4b. At the peptide level, all members of this family possess multiple, tandemly arranged repeated structures called short consensus repeats (SCRs). Each SCR is 65–70 amino acids long with 11–14 conserved amino acid residues. The composition and number of SCRs each of these members contain vary, but structurally and functionally they form the foundation by which this family is based. The genes encoding H, CR1, CR2, DAF, C4 bp, and MCP are found within an 800-kb genomic segment on the long arm of chromosome 1.
Another group of complement genes that are linked together are C2, factor B, and C4 that all map within the major histocompatibility complex of both humans and mice. In the human, these complement genes, often called the Class III genes, map between the Class II HLA-DR and Class I B loci of chromosome 6. Similar to the Class I and Class II genes, the Class III genes are polymorphic as multiple alleles exist for each of these genes.
Complement components C3, C4, and C5 constitute a structurally homologous family of proteins that includes a2-macroglobulin and pregnancy zonal protein. All except C5 are characterized by the presence of an interal thioester bond allowing these proteins to covalently bind with cell surfaces or other proteins. However, unlike the RCA family, C3, C4, and C5 reside on separate chromosomes (Table 1).
14. Pathologies Related to the Complement System
The complement cascade is a potent and powerful system whose function is the destruction of target cells and infectious agents. Unwanted deposition of complement as well as the production of significant quantities of inflammatory mediators can result in substantial damage to normal healthy tissues. The regulatory mechanism of the MAC and alternative and classical pathways establishes a fine balance between activation and inhibition so as to protect autologous cells but allow the destruction of foreign agents. Deficiencies in nearly any component of the complement system compromise the health of an individual by tipping the balance one way or another. In a general sense, complement deficiencies can be due to the absence of a component due to structural defect in the gene, or the production of a component that is incapable of functioning in its specific role in the cascade. Table 2 summarizes complement deficiencies, the resulting abnormalities, and associated pathologies.
Table 2. Complement Deficiencies and Associated Clinical Abnormalities


Deficiencies in alternative and classical pathway components usually manifest clinically in recurrent bacterial infections. The most serious deficiencies are those in C3. Given its role in opsinization, phagocytosis, and lysis of bacteria, homozygous deficiencies in C3 often prove fatal. In contrast, C9 deficient individuals cannot effectively generate MAC formation, yet these patients have minimal or no associated pathology as the addition of C8 to the C5b-7 complex can result in osmotic lysis of target cells. Individuals deficient in the early classical pathway components (C1, C2, and C4) are impaired in their ability to solubilize and clear immune complexes, resulting in local inflammation associated with autoimmune diseases such as systemic lupus erythematosus (SLE.(
Patients that are deficient in the terminal complement components (C5 through C9) have impaired ability to assemble the MAC. Of particular interest is that these patients seem to have a propensity for Neisseria bacterial infections suggesting that cytolysis may be the major defense mechanism against these bacteria.
Deficiencies in the soluble and membrane bound regulatory components of complement result in abnormal activation and deposition of complement. Defects in C1INH results in a condition called hereditary angioneurotic edema (HANE), in which the intermittent accumulation of edema ) swelling) in the skin and mucosa occurs. The exact mechanism of how the edema occurs is not known; however, without C1INH to inhibit non-specific C1 activation, C2 and C4 are readily cleaved and their by products are implicated. Deficiencies in the expression of phosphytidylinositol-linked membrane proteins result in a condition called paroxysmal nocturnal hemoglobinuria (PNH), where patients suffer from recurrent bouts of intravascular hemolysis. As DAF and CD59 are PI-linked, they also serve to inhibit C3 convertase formation. However, erythrocytes are especially sensitive to lysis as they do not possess membrane bound form of CD59 as other cell types do.
15. Summary
The proteins of the complement system consists of at least 20 chemically and immunologically distinct plasma proteins that can dynamically interact with one another in a highly regulated manner. The complement system can be activated by using either of two converging initiation pathways: the classical pathway, which is activated by antibody–antigen complexes; and the alternative pathway, which is activated by the surface of invading organisms. This interaction of proteins results in (1) the lysis of target cells and bacteria, (2) the binding and clearance of immune complexes throughout the body, (3) the binding of complement based proteins to foreign particles and subsequently cleared by phagocytosis, and (4) the generation of inflammatory mediators to enhance the humoral and cellular immune response. The activation of complement is tightly regulated so as to protect host cells from nonspecific attack deposition while allowing the destruction of foreign agents. Deficiencies in complement components of either pathway or in regulatory components are associated with recurrent bacterial infections and autoimmune diseases.
16. Acknowledgments
This is publication 133-IMM from the Institute of Molecular Medicine for the Prevention of Human Diseases, University of Texas-Houston Health Science Center. This work was supported by GM56050 (DLH) and AI25011 (RAW). This article is dedicated to the memory of Dr. Hans Müller-Eberhard.
References
A. K. Abbas, A. H. Lichtman, and J. S. Pober (1994) Cellular and Molecular Immunology, 2nd ed., Saunders, Philadelphia.
H. Müller-Eberhard (1986) The membrane attack complex, Annu. Rev. Immunol. 4, 503–528.
H. Müller-Eberhard (1988) Molecular organization and function of the complement system, Annu. Rev. Biochem. 57, 321–347.
R. A. Wetsel and H. R. Colten (1990) in Inheritance of Kidney and Urinary Tract Diseases, A. Spitzer and E. D. Avner, eds., Kluwer Academic, Chapter "18", pp. 401–429.
G. D. Ross, ed. (1986) Immunobiology of the Complement System, Academic Press, San Diego, CA.
K. Rother and G. O. Till, eds. (1988) The Complement System, Springer-Verlag, New York.
K. Rother and U. Rother, eds. (1986) Hereditary and Acquired Complement Deficiencies in Animals and Man, Progress in Allergy, Vol. 39, Karger.

|
|
|
|
التوتر والسرطان.. علماء يحذرون من "صلة خطيرة"
|
|
|
|
|
|
|
مرآة السيارة: مدى دقة عكسها للصورة الصحيحة
|
|
|
|
|
|
|
نحو شراكة وطنية متكاملة.. الأمين العام للعتبة الحسينية يبحث مع وكيل وزارة الخارجية آفاق التعاون المؤسسي
|
|
|