آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Collagenopathies |


|
|
Read More
Date: 7-9-2021
Date: 10-9-2021
Date: 21-11-2021
|
Collagenopathies
Defects in any one of the many steps in collagen fiber synthesis can result in a genetic disease involving an inability of collagen to form fibers properly and, therefore, an inability to provide tissues with the needed tensile strength normally provided by collagen. More than 1,000 mutations have been identified in 23 genes coding for 13 of the collagen types. The following are examples of diseases (collagenopathies) that are the result of defective collagen synthesis.
1. Ehlers-Danlos syndrome: Ehlers-Danlos syndrome (EDS) is a heterogeneous group of connective tissue disorders that result from heritable defects in the metabolism of fibrillar collagen molecules. EDS can be caused by a deficiency of collagen-processing enzymes (for example, lysyl hydroxylase or N-procollagen peptidase) or from mutations in the amino acid sequences of collagen types I, III, and V.
The classic form of EDS, caused by defects in type V collagen, is characterized by skin extensibility and fragility and joint hypermobility (Fig. 1). The vascular form, due to defects in type III collagen, is the most serious form of EDS because it is associated with potentially lethal arterial rupture. [Note: The classic and vascular forms show autosomaldominant inheritance.] Collagen that contains mutant chains may have altered structure, secretion, or distribution, and it frequently is degraded. [Note: Incorporation of just one mutant chain may result in degradation of the triple helix. This is known as a dominant-negative effect.]
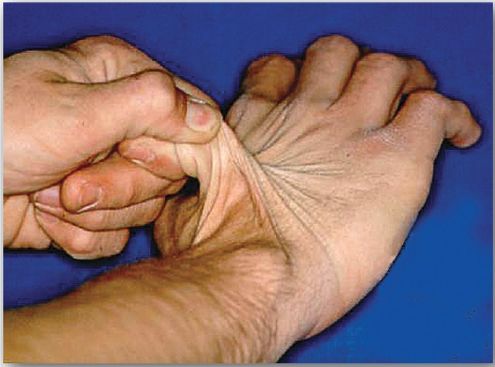
Figure 1 Stretchy skin of classic Ehlers-Danlos syndrome.
2. Osteogenesis imperfecta: This syndrome, known as “brittle bone disease,” is a genetic disorder of bone fragility characterized by bones that fracture easily, with minor or no trauma (Fig. 2). Over 80% of cases of osteogenesis imperfecta (OI) are caused by dominant mutations to the genes that encode the α1 or α2 chains in type I collagen. The most common mutations cause the replacement of glycine (in –Gly–X–Y–) by amino acids with bulky side chains. The resultant structurally abnormal α chains prevent the formation of the required triple-helical conformation.
Phenotypic severity ranges from mild to lethal. Type I OI, the most common form, is characterized by mild bone fragility, hearing loss, and blue sclerae. Type II, the most severe form, is typically lethal in the perinatal period as a result of pulmonary complications. In utero fractures are seen (see Fig. 2). Type III is also a severe form and is characterized by multiple fractures at birth, short stature, spinal curvature leading to a humped-back (kyphotic) appearance, and blue sclerae. Dentinogenesis imperfecta, a disorder of tooth development, may be seen in OI.
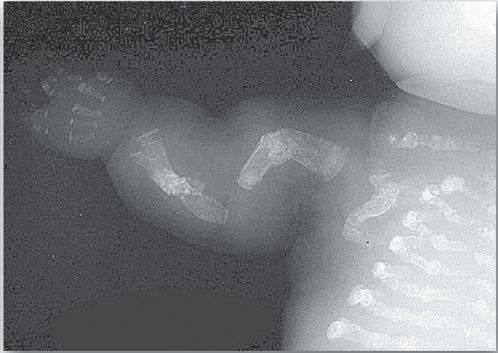
Figure 2: Lethal form (type II) of osteogenesis imperfecta in which the fractures appear in utero, as revealed by this radiograph of a stillborn fetus.

|
|
|
|
دراسة يابانية لتقليل مخاطر أمراض المواليد منخفضي الوزن
|
|
|
|
|
|
|
اكتشاف أكبر مرجان في العالم قبالة سواحل جزر سليمان
|
|
|
|
|
|
|
اتحاد كليات الطب الملكية البريطانية يشيد بالمستوى العلمي لطلبة جامعة العميد وبيئتها التعليمية
|
|
|