آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Chromatin |


|
|
Read More
Date: 15-3-2021
Date: 2025-03-22
Date: 5-3-2021
|
Chromatin
Chromatin is the complex of histones and DNA, associated with smaller amounts of other proteins, into which the chromosomal DNA of all eukaryotes is organized. As well as being a means of packing DNA in an orderly fashion, chromatin structure plays a crucial role in the regulation of gene expression, both activation and repression. Three books contain much useful information about chromatin structure and function (1-3); for the most up-to-date information in many areas of this rapidly moving field, the reader will be referred to recent articles and reviews.
1. Overview
Chromatin has a beaded appearance in the electron microscope at low ionic strength, arising from a regularly repeating structure (4). The repeating unit is the nucleosome, which contains ~166 to 240 bp of DNA, of which ~166 bp is wound in two left-handed superhelical turns around an octameric complex of the four core histones (H3, H4, H2A, and H2B) and stabilized by one molecule of histone H1; the nucleosome also includes a variable length of linker DNA (~0 to 76 bp( that connects neighboring nucleosomes. The octamer is organized as a central H32H42 tetramer, which has a rather flat, twisted horseshoe shape, flanked above and below by an H2A–H2B dimer. The details of the organization of histone and DNA in the core of the nucleosome are revealed in the high-resolution structure of the 146-bp nucleosome core particle (which contains no linker DNA and no H1) recently determined by X-ray crystallography (5). The globular domain of histone H1 binds asymmetrically, bridging a point near the dyad and an entering or exiting DNA double helix (6); the basic C-terminal domain interacts with the linker DNA, partially neutralizing its charge. The array of nucleosomes is known as a nucleosome filament, or the 10-nm filament (10 nm being the diameter of the nucleosome). The amount of DNA contained in the repeating unit, which reflects the length of linker DNA, is determined from the sizes of the DNA in the chromatin fragments liberated by incomplete digestion with micrococcal nuclease (Staphylococcal nuclease), which cuts in the linker. A series of fragments, usually visualized as a “ladder” of bands in gel electrophoresis, occurs at multiples of the unit repeat size. This is commonly about 200 bp (when there is a linker length of 200 – 166 = 34 bp), but it may be as long as ~240 bp ) in sea urchin sperm), or as short as ~166 bp (essentially no linker DNA) in yeast and in mammalian cerebral cortex neurons (the glial cells in the same tissue have a repeat length of about ~200 bp). The repeat length measured in this way is the average for the tissue or cell population under examination; there will be local variations about this mean. Packaging of the DNA in nucleosomes achieves about a sixfold length compaction of the DNA, and further packing is achieved by salt-dependent, histone H1-assisted folding of the 10-nm nucleosome filament into a 30-nm filament. Much evidence supports a simple helical coiling of the nucleosome filament into a “solenoid” with six nucleosomes per turn, but there are other models. Most of the chromatin in the cell is in the form of a 30-nm filament throughout most of the cell cycle, and at interphase appears to be looped onto the nuclear matrix; at mitosis, the 30-nm filament is subjected to further levels of folding to give the highly dense metaphase chromosome in which the overall length compaction of the DNA is about 10,000-fold.
The 30-nm filament is not a suitable template for transcription by the large eukaryotic RNA polymerases and clearly has to be unfolded first; formation of an “open” (“transcriptionally competent”) chromatin state is an essential prerequisite for transcription. This involves acetylation of the basic N-terminal regions of the core histones, which appear to play a role in chromatin folding, and some depletion of histone H1. It has recently become clear that acetylases are targeted to particular regions of the chromosome through recruitment, directly or indirectly, by gene regulatory proteins bound to specific DNA sequences at promoters or upstream activating sequences. Cells appear to contain many acetylase (and deacetylase) activities, which function as components of multiprotein complexes. Even in the 10-nm filament form, nucleosomes may act as blocks to the formation of transcription initiation complexes by occluding promoters, which may include strategically positioned nucleosomes. It is now clear that the cell has specialized energy-dependent mechanisms that are required for “remodeling” some promoters, so that they become accessible to transcription factors, as well as mechanisms for facilitating RNA chain elongation. These mechanisms also involve multiprotein complexes, which may have subunits in common with acetylases and deacetylases. Chromatin is thus both an effective means of packing and the target for an integrated network of machines that modify it. There is a wealth of genetic evidence showing that the growing list of factors that regulate transcription includes both bona fide chromatin components (eg, histones) and proteins that modify chromatin (eg, acetyltransferases and deacetylases). An unexpected discovery was that a structural motif found in all the core histones is also found in a number of components of the transcription initiation factor TFIID, as well as in some histone acetyltransferase complexes, suggesting a structural and evolutionary link between chromatin and the assemblies that act upon it. Another surprising finding has been that histone H1 may have a specific gene regulatory role, in addition to its role in packaging and as a general repressor.
2. Nucleosome Positioning and Mobility
Histones package the whole of the genome and therefore appear to be largely indifferent to DNA sequence. Locally, the position of a nucleosome is determined by the underlying DNA sequence and, in particular (because the DNA, which is normally rigid, is bent around the octamer surface), by the local bendability of DNA, which is sequence-dependent. Analysis of the DNA sequences of bulk nucleosome core particles shows that A and T di- and trinucleotides are likely to occur where the minor groove faces inward, and G and C di- and trinucleotides where the minor groove faces out. This is because the minor groove of AT-rich DNA is naturally narrow and so, by having A and T where the minor groove faces in, the compression of the groove that occurs on wrapping the DNA around the octamer is readily accommodated (7). The dinucleotide periodicity is on average 10.2 bp, exactly the average structural periodicity given by the X-ray crystal structure of the nucleosome core particle. The helical periodicity in solution is 10.6 bp, so the DNA is overtwisted in the nucleosome, presumably to achieve the match between the structural and sequence periodicities that underlie the rules that govern rotational positioning of the octamer with respect to the DNA sequence (the orientation of a face of the duplex with respect to the histone octamer surface). This is thus determined by the local bendability (flexibility toward curvature) of the DNA, which is determined by the properties of the individual base steps. A rotationally positioned duplex shows a characteristic 10-nucleotide pattern of nicking by DNase I, as each strand rises from the surface about every 10 nucleotides. Translational positioning, on the other hand (the basis of which is not well understood), refers to the choice of a particular stretch of DNA by the histone octamer, rather than other stretches of the same length translated forward or backward along the DNA by about 10 bp, which would allow the same rotational setting. The abundance of AA/TT dinucleotides separated by roughly 10 bp led to the construction of fragments with alternating A/T and C/C sequences [(A or T)3NN(G or C)3NN]n—the so-called TG pentamer—that indeed formed very stable nucleosomes (8). Selection of naturally occurring 146-bp DNA sequences with a high affinity for the histone octamer revealed (in addition to the A/T, G/C periodicity) that sequences with repeated TATAAAACGCC motifs (“phased TATA” sequences) formed nucleosomes that were even more stable, suggesting that high-affinity binding is aided by flexible sequences (9); selection from longer (220 bp) synthetic DNA sequences also revealed a signal (CTAG) that favored high-affinity binding (10). Such sequences, even if they occurred infrequently in the genome, could have important implications for chromatin organization and regulation, as could sequences that are refractory to nucleosome formation (eg, TGGA repeats identified in a negative-selection approach (11) . It seems likely that the signals discovered for rotational positioning in vitro (7) also function in vivo, as revealed by analysis of the complete genome sequences of the yeast Saccharomyces cerevisiae and the nematode Caenorhabditis elegans (12), where dinucleotide periodicities are compatible with nucleosomal constraints. Analysis of 168-bp chicken erythrocyte chromatosome sequences [ie, containing H5(H1] shows that the statistical preferences of the octamers for particular sequences are slightly modulated (13), presumably in order to optimize the stability of the octamer–DNA–H1 ternary complex. This would be reflected in slight differences in translational positions of nucleosomes in the presence of H5(H1), which might have functional consequences (eg, in exposing or revealing short DNA sequences in an H1-dependent manner). An additional feature of chromatosomal DNA is the frequent occurrence of AGGA within half a double helical turn of one terminus, imposing asymmetry on the chromatosome. Whether this is related to the asymmetric binding of H1 to the nucleosome remains to be seen. The rules for nucleosome organization on DNA sequences have recently been reviewed (14).
Although histones can, in general, package DNA essentially irrespective of sequence, but capitalize on the local bendability of DNA to promote a better fit on the octamer surface, some DNA sequences have a particularly high affinity for the octamer, leading to a “positioned nucleosome” in which a particular translational position is preferred. Nucleosome reconstitution experiments show that the H3–H4 tetramer alone is sufficient to confer positioning on a defined DNA sequence (15, 16). Positioned nucleosomes have been identified in vivo by nucleosome mapping techniques and in vitro in nucleosome reconstitution experiments on defined DNA sequences. The rules for nucleosome positioning in vivo are not well understood: When the TG pentamer was introduced into yeast, it appeared to exclude nucleosomes rather than provide optimal positioning (17), showing that a strong rotational setting is not sufficient for positioning in vivo. Positioned nucleosomes may act as a boundary (18) and determine the positions of several neighboring nucleosomes on either side. More than one positioned nucleosome separated at less than a nucleosome's length of DNA would serve to keep the region clear of histones (eg, at promoters) for the binding of sequence-specific proteins with various gene regulatory or structural roles. At the inducible yeast PHO5 promoter, events leading to promoter remodeling and transcription of the PHO5 gene, which encodes a phosphatase, are initiated in response to low phosphate levels by binding of the transcription factor PHO4 to a site between two positioned nucleosomes in a set of four over the promoter (19). Positioned arrays of nucleosomes, essential for the functioning of the promoter, are also found in many other cases—for example, the mouse mammary tumor virus (MMTV) major late promoter (20) (see also Ref. 2). It has recently become apparent (21) that nucleosome positioning is the probable explanation for the differential regulation of Xenopus oocyte and somatic 5S genes in vivo (22), where the oocyte genes are repressed and the somatic genes remain active when H1 accumulates after the mid-blastula transition. Sequence differences between the two types of gene mean that the oocyte nucleosome occludes the TFIIIA binding site and binds H1 preferentially (so the gene is turned off), whereas the converse is broadly true for the somatic gene, which remains active.
A nucleosome core can be reconstituted on to DNA in vitro by dialysis of an equimolar mixture of the histone octamer (or H3–H4 tetramers and H2A–H2B dimers) and DNA from high ionic strength (eg, 2 M NaCl) to low ionic strength. Even in cases where there are strong nucleosome positioning signals in the DNA, octamers on different fragments will occupy a population of less favorable minor translational positions in addition to a major position, all with the same rotational setting of the DNA. For example, on the Xenopus borealis somatic 5S rRNA gene, which is regarded as a strong positioning sequence, octamers can occupy 10 different positions (six being predominant), all related to each other by the 10 bp helical periodicity of the DNA, which must be caused by a strong rotational signal in the DNA sequence (21). The occupancy of less favorable octamer positions on a DNA sequence is higher at low temperatures (eg, 4°C), when the octamers are trapped, than if the temperature is then shifted to 37° (23). The change in positions populated demonstrates that nucleosomes have an intrinsic mobility, which could be useful, for example, in remodeling of promoters. In the presence of H1, nucleosome mobility is greatly reduced (24, 25); conversely, loss of H1 might enable nucleosomes to move, for example, to expose binding sites for gene regulatory proteins.
3. Higher-Order Structure: The 30-nm Filament
The 10-nm nucleosome filament, which represents the first level of chromatin structure, is the form in which chromatin exists at low ionic strength (eg, >20 mM) in vitro. As the ionic strength is increased, the 10-nm filament condenses into a 30-nm (diameter) filament, visible by electron microscopy. Most of the chromatin in the nucleus is in this form. The folding is a consequence of ionic-strength induced screening of the residual negative charge on the linker DNA (26), much of the charge having already been neutralized by the basic tail(s) of histone H1 and, probably, the basic N-terminal tails of at least some of the core histones, and results in new interactions (eg, nucleosome-nucleosome) within the higher-order structure.
Various models have been proposed for the 30-nm filament (27), some of which are shown in Figure 1. All agree on a radial distribution of nucleosomes with their faces parallel to the filament axis, but they differ fundamentally in the path taken by the linker DNA, whether bent, curved, or straight. The evidence on this point is conflicting. The evidence for the various models has been extensively reviewed (28-31). Much evidence supports the original “solenoid” model (32), which proposes a simple helical coiling of the nucleosome filament, with about six nucleosomes per turn at physiological ionic strength, and necessarily bent linker DNA (Fig. 1a). [The details are easily modified to accommodate the asymmetric, rather than symmetric, location of the globular domain of H1 near the dyad (6); see text above.] The solenoid places the dyads of the nucleosomes, histone H1, and the linker DNA between nucleosomes, on the inside of the solenoid; H1 has indeed been shown to be located within the 30-nm filament (33). However, other models have been proposed, such as the helical-ribbon model (34, 35) (Fig. 1b), which invokes straight linkers and is based on the zigzag appearance of the nucleosome filament in electron micrographs, and these too would be compatible with an internal location of H1. The coiled linker model (36), like that shown in Fig. 1c, which is related to the solenoid, is not necessarily compatible, because the dyads of the nucleosomes, and hence H1, are alternately inside and outside the solenoid for some linker lengths (eg, in chicken erythrocyte chromatin).
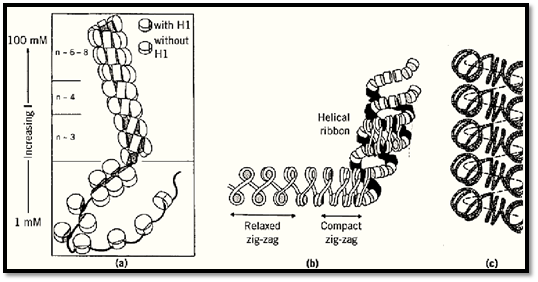
Figure 1. Some models of chromatin higher-order structure. The 30 filament is shown as (a) a solenoid (32), (b) a helica ribbon (34), and (c) as a helical conformation with coiled linkers (36). In (a) and (b) the most condensed structures are shown at the top, and the extended structures present at low ionic strength at the bottom. (From Refs. 32, 34, and 36, respectively, with permission.)
Whatever the model that best describes the spatial relationships between nucleosomes in the higher-order structure, the 30-nm filament in the cell nucleus is unlikely to be completely regular, partly because of local heterogeneity in linker length along the filament within the average value, partly due to the presence of different histone variants or subtypes, and so on. The 30-nm filament is also a dynamic structure that undergoes thermal “breathing”, and this property is probably central to gene regulation through control of chromatin folding. The stability of the 30-nm filament is relevant to the ease with which it may be unpackaged for transcription and appears to be increased by special variants of H1 (such as H5 and spH1 in the condensed chromatin of nucleated erythrocytes and sea urchin sperm, respectively) (37), and it is probably decreased in regions of transcriptionally competent chromatin by (partial) loss of H1 (eg, Ref. 38) and by acetylation of the core histone N-terminal tails.
4. Chromatin and Transcription
In eukaryotes, transcription works in a chromatin context (39, 40). Some genes are active in all cell types, whereas others are active only in specific cell types. Active genes in higher eukaryotes have a chromatin structure characterized by a greater sensitivity (7- to 10-fold) to the endonuclease DNase I than the bulk of the chromatin in the nucleus (1, 2). This state precedes active transcription and marks “transcriptional competence”; it is the first step in control of transcriptional activity. DNase I sensitivity is probably due to (partial) relaxation of the 30-nm filament toward the 10-nm filament state, which is probably facilitated by partial loss of linker histones and acetylation of the core histone N-terminal tails (41). DNase I sensitivity associated with a particular gene extends well beyond the coding region and encompasses an entire chromosomal domain (eg, tens of kilobases) and includes all the regulatory elements (eg, enhancers) for the genes (2) . The boundaries of these chromosomal domains have been characterized in only a few cases, and no common feature is apparent. In at least some instances, the boundaries act as “insulators”, preserving the structural and functional autonomy of the domain and also blocking the influence of regulatory elements from the outside (42). Short localized regions, termed DNase I hypersensitive sites (typically a few hundred base pairs), in the genome show a much higher sensitivity to DNase I than described above (43). These are due to the absence of a canonical nucleosome and the presence of gene regulatory (sequence-specific) proteins. They often occur within a domain of general DNase I sensitivity, coinciding with promoters or enhancers, and may also occur at the boundaries of chromosomal domains. Hypersensitive sites at promoters may be constitutive or inducible, depending on the gene, and are often flanked by “positioned nucleosomes”.
The presence of a nucleosome at a promoter generally inactivates it (39). Promoters therefore either have to be kept clear of nucleosomes, or one or more nucleosomes has to be perturbed or disrupted to permit assembly of a preinitiation complex and recruitment of RNA polymerase. Promoters may be kept clear either by flanking positioned nucleosomes or by the presence of sequence-specific proteins that bind during replication—for example, the Grf2 protein in yeast, which binds to an extended region of DNA and precludes nucleosome formation (39). The promoters that are packaged in nucleosomes have to be opened up (“remodeled”) in order to permit assembly of a preinitiation complex. The nature and extent of the disruption required to open up a promoter is unclear and may be different in different cases, perhaps depending on where the factor-binding sequences are within a nucleosome (eg, whether they can be exposed simply by loss of H1 or by loss of an H2A-H2B dimer, or whether they also require disruption of the central tetramer in the core of the nucleosome; see Ref. 2). It does not appear to result in complete histone loss. Promoter remodeling and disruption in vivo appears to require the action of energy-dependent “chromatin remodeling machines” which may be recruited to particular promoters in response to the initial signal for activation, and possibly also histone acetyltransferases (see Histone Acetylation).
In vitro many transcription factors have been shown to be able to can bind to their cognate sites on the surface of a nucleosomes, albeit with lower affinity than to free DNA. The effects can be accounted for by transient “site exposure” as the interactions between the octamer and DNA fluctuate (44); this model predicts that binding of factors to multiple juxtaposed binding sites on the same nucleosome will be cooperative (45), as indeed already observed. Binding occurs without global displacement of the DNA, which will remain anchored at sites not bound by transcription factors. Consistent with this, a recent report shows that the erythroid transcription factor GATA-1 causes extensive disruption of histone–DNA contacts in nucleosome core particles containing GATA-1 sites, except over 50 bp around the dyad that serves to anchor the DNA (46). The complex is stable in solution, and the disruption is reversed on removal of GATA-1. The inherent flexibility of the nucleosome, and the fact that many promoters have clusters of factor-binding sites, suggests that this mechanism of gaining access to binding sites within nucleosomes at promoters could have physiological relevance.
At some inducible promoters, the binding site for the transcription factor that initiates the remodeling events is already exposed, in the linker between two nucleosomes in a short positioned array, rather than within a nucleosome. This is the situation at the PHO5 promoter in yeast (19). Activation of the promoter in response to low phosphate relies on the accessibility in the uninduced state of a (weak( binding site for a transcription factor (Pho4) in the 70-bp gap between two of the four positioned nucleosomes at the promoter (Fig. 2). Binding of Pho4 leads to disruption of two nucleosomes on either side of the binding site and exposure of a second, stronger Pho4 binding site, as well as binding sites for another transcription factor, Pho2, which binds cooperatively with Pho4. The nature of the disruption is not clear but is reflected in the accessibility to a restriction enzyme of a site normally incorporated into a nucleosome and probably involves a “remodeling complex”. In other cases, a nucleosome at a promoter may be beneficial or even essential for activation of transcription. In the mouse mammary tumor virus (MMTV) long terminal repeat (LTR) promoter, six positioned nucleosomes place two glucocorticoid response elements in rotational positions on one of the nucleosomes, such that they are accessible to glucocorticoid receptor binding in response to hormone; the two sites are also in close proximity on one face of the nucleosome. The binding site for the transcription factor NF1 is occluded until the nucleosomes are disrupted or perturbed in response to receptor binding (20). In a slightly different scenario, wrapping DNA around a nucleosome may bring into proximity two sites outside the nucleosome core, which are separated by about a nucleosome repeat length of DNA. There are several instances of this (2)— for example, the Drosophila hsp26 promoter (47), where a critical nucleosome upstream of the start site of transcription performs such a scaffolding role and brings binding sites for heat shock transcription factors into proximity with each other and with TFIID, to create a preset promoter, with bound RNA polymerase, waiting only for the binding of heat shock transcription factor to induce it.
The positioned nucleosome is flanked by binding sites for the GAGA protein, which appears to play a crucial role in the architecture of the promoter, by helping to determine the position of the nucleosome (2).
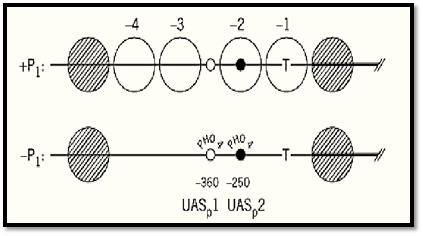
Figure 2. The yeast PHO5 promoter: chromatin structure in the uninduced (+Pi) and induced (–Pi) states. Large open circles (–1 to –4): the four positioned nucleosomes that are disrupted and become “transparent” upon induction. Small circles: Pho4 binding sites [UASp1 (open) and UASp2 (filled)]. T: TATA box. The thick horizontal line marks the beginning of the coding sequence. From Ref. 75, with permission.
In contrast to initiation, transcriptional elongation can proceed through nucleosomes in vivo. In a model system using SP6 polymerase and a single nucleosome, the histone octamer is displaced during elongation (39) and then rebinds upstream of its initial position; the mechanism proposed is transferred from in front of the advancing polymerase to behind it, via a DNA loop that is displaced (48). However, no such studies have yet been carried out with the large eukaryotic RNA polymerases. The indications are that a reconstituted nucleosome array presents a block to elongation by RNA polymerase II, which may be relieved by an energy-dependent mechanism.
5. Chromatin Remodeling Machines
Exciting discoveries in the last couple of years have revealed several multiprotein complexes ) molecular masses ranging from 0.5 to 2 MDa) in Drosophila, yeast, and human cells that are able to remodel repressive chromatin in an ATP-dependent manner to permit transcription initiation at promoters in vitro. These include three complexes derived from a Drosophila embryo extract: NURF (nucleosome remodeling factor), CHRAC (chromatin accessibility complex) and ACF (ATP-dependent chromatin assembly and remodeling factor); the Saccharomyces cerevisiae SWI-SNF ( switching-sucrose nonfermenting) and related RSC (remodeling the structure of chromatin( complexes; and SWI/SNF-related complexes in human cells and Drosophila. They have been comprehensively discussed in a number of recent reviews (49-53) that cite the original literature. They all contain an ATPase activity; in the case of the three Drosophila complexes, this is the same ISWI (imitation SWI2) subunit. The various complexes were purified initially using a range of functional assays to follow the purification, and they appear to be functionally and mechanistically distinct; they are likely to differ in their modes of nucleosomal perturbation. NURF (four protein subunits) was purified as a factor required for ATP-dependent remodeling of chromatin templates so that the GAGA protein is able to bind (GAGA binds to GA-rich sites in several Drosophila heat-shock promoters, such as the hsp26 promoter, already mentioned); and it facilitates transcriptional activation. Interestingly, the 55-kDa subunit of NURF (which contains “WD repeats,” which are protein-interaction motifs) is also found in the Drosophila chromatin assembly factor dCAF-1 (54), and human homologues are components of histone acetylase and deacetylase complexes. CHRAC (five subunits) was purified as a factor that generated ATP-dependent accessibility of sites in a chromatin template, reconstituted in vitro, to restriction enzymes. It is also able to generate a regular array from an irregular array of nucleosomes and has been described as an energy-dependent nucleosome spacing factor; it may therefore play a role in chromatin assembly where newly assembled nucleosomes are initially deposited in an irregular array that “matures” to give a regular spacing), although a spacing activity might, of course, also promote transcription factor binding. The third Drosophila complex, ACF (four subunits), was also purified as an ATP-dependent nucleosome spacing factor in a fractionated system and plays a role in nucleosome assembly; it can also mediate transcription-factor-mediated chromatin disruption in much the same way as NURF. SWI/SNF (9 to 12 subunits) facilitates transcription factor binding, and both SWI/SNF and RSC (15 subunits) disrupt the repeating 10-nucleotide cleavage pattern given by DNase I on mononucleosomes, showing disruption of the rotational setting of the DNA on the octamer surface. In yeast, the SWI/SNF complex is not abundant and not essential for viability, whereas the RSC complex is abundant and essential. SWI/SNF appears to be necessary for the transcription of only a small subset of genes, and in an in vitro system the dependence of transactivation on SWI/SNF activity is much reduced if there are multiple binding sites for the activator on the same nucleosome (55). It is possible that in vivo the SWI/SNF complex is needed only for remodeling of weak promoters. Altered forms of mononucleosomes generated by both SWI/SNF (56, 57) and RSC (58) complexes have recently been reported. They retain a full complement of histones and have altered physical properties that would be consistent with either a heavily modified mononucleosome or a dinucleosome. The SWI/SNF products have a higher affinity for transcription factors than unmodified nucleosomes. Much remains to be done to (a) elucidate the mechanisms by which the various chromatin remodeling machines act on chromatin, (b) define the roles of the various subunits, and (c) establish which mechanisms, in vivo, play a role in promoter disruption for transcription initiation, and whether others are primarily involved in reversing such disruption or in nucleosome assembly, and so on. The relationship between these complexes and the large acetylase, and deacetylase, complexes also remains to be clarified, although the indications are that they cooperate in the remodeling of certain promoters.
The foregoing account has dealt solely with complexes that disrupt chromatin at promoters, allowing transcription initiation complexes to form. The transcriptional machinery also confronts nucleosomes during RNA chain elongation. Experiments on a reconstituted array of nucleosome cores show that the passage of eukaryotic RNA polymerase II is blocked by a nucleosome core and that a heterodimeric protein isolated from human cells will facilitate RNA chain elongation in a chromatin context. The protein has been designated FACT (facilitates chromatin transcription) (59) and appears to be a DNA-binding protein, one subunit being an HMG-box protein. Little is yet known about it how it might work, but its action is not ATP-dependent. The in vitro assays show a rather modest extension of the transcript (50 to 150 nucleotides) before FACT stalls, and it is not yet clear whether FACT is designed to work only on the promoter-proximal nucleosome or needs cofactors for its normal action, or whether the modest extension is merely due to a technical limitation of the experiment. The chromatin template contains closely packed nucleosome cores (no linker DNA, no H1), and if octamer transfer via a loop of DNA is required, as demonstrated in model systems (48 (, this may not be possible here.
6. Chromatin and Repression
There are hierarchies of repression, through chromatin structure, as there are of transcriptional activation. The most basic level, promoter occlusion by a nucleosome, was discussed above. Superimposed on this is repression through folding of the nucleosome filament into a 30-nm filament (whether this occurs in yeast is unclear). The higher-order structure is stabilized by a hypoacetylated state of the N-terminal tails of the core histones. In organisms other than yeast and Drosophila, DNA methylation is known to contribute to gene repression through chromatin structure, and a 5-methylcytosine-binding protein (MeCP2) has recently been reported to recruit deacetylases (60, 61). This could favor a more stable 30-nm filament, which might be further stabilized by the tighter binding of H1 to methylated DNA (62). The 30-nm filament is not a template for transcription, and in some cases, 30-nm filament stabilization (eg, by specialized linker histone variants) and packing in the nucleus may be at least major players in general repression. This may be the situation in mature nucleated erythrocytes (eg, of birds, fish, and amphibia), where H1 is largely replaced by histone H5 and where there is a global and terminal shutdown of transcription; and in sea-urchin sperm, where highlycondensed chromatin is linked to the presence of sperm-specific H1. Other more complex mechanisms of repression also exist, however, resulting in heterochromatin. The essence of these mechanisms—although they differ in many respects in, for example, yeast and Drosophila, where they have been extensively studied—is the assembly, over particular chromosomal regions, of repressive multiprotein complexes that interact with chromatin. In the case of Drosophila at least, the chromatin is probably in the form of the 30-nm filament, and the mechanisms add an additional layer of repression for genes whose permanent inactivation is crucial and where inappropriate expression cannot be tolerated.
Yeast chromosomes are too small to allow heterochromatin to be seen cytologically, but nonetheless some chromosomal regions show many of the features of heterochromatin in more complex eukaryotes (replication late in S phase, localization at the nuclear envelope, and position effects on gene expression that are inherited epigenetically). In Saccharomyces cerevisiae, such regions occur at the silent mating type loci, HML and HMR, and at telomeres (the ends of the chromosomes; genes placed here are repressed) and have been extensively studied by genetic analysis (63). In each case, a multiprotein complex of Sir2, Sir3, and Sir4 proteins (Sir = silentinformationregulator information regulator) interacts with the N-terminal regions of histones H3 and H4. At the telomeres, which contain the sequence [C1–3A]n, the silencers consist of multiple binding sites for RAP1 (repressor activator protein 1), whereas silencers at HML and HMR consist of binding sites for ABF1 (ARS-binding factor 1), the origin recognition complex (ORC) and RAP1. Residues 16 to 29 of H4 are required for interaction with Sir3 and Sir4 and for silencing, presumably by providing interactions necessary for stabilization of the multiprotein complex. In addition, although the core histones at the silenced loci in yeast are hypoacetylated, there is a specific requirement for Lys12 in acetylated form, possibly for interaction with Sir3 (63). In the current model for telomeric repression, RAP1 binds to the telomeric repeats, initiating polymerization of Sir3/Sir4 over the adjacent chromatin domain, the binding of the Sir proteins being mediated by interaction with the H3 and H4 N-termini, possibly stabilized by acetylation of H4 at Lys12. Other repressive mechanisms in yeast also involve the N-terminal tails of H3 and H4 (64).
Extensive genetic studies of heterochromatin have also been carried out in Drosophila. Genes brought into juxtaposition with heterochromatin (eg, at centromeres) may be silenced in a subset of cells that normally express the gene—an effect known as position effect variegation (PEV) (2). Chromatin structure is altered in variegating genes, as shown by the properties of the hsp26 transgene inserted at various chromosomal locations; the heterochromatic hsp26 gene has a noninducible promoter and is packaged in an unusually regular nucleosomal array (65). Modifiers of PEV, identified genetically, are either (a) structural proteins that are components of multimeric protein complexes that interact with chromatin or (b) proteins that could play indirect roles in regulating the formation of chromatin—for example, by modifying histones or DNA. One such protein is the histone deacetylase RPD3. However, mutations in this protein that increase acetylation increase PEV, rather than decreasing it, as might be expected; this is probably because of the requirement for acetylation at Lys12, as in yeast heterochromatin, against a background of general histone hypoacetylation. The multiprotein complexes that affect PEV, and presumably stabilize chromatin higher-order structure, contain (a) HP1 (heterochromatin protein 1), which binds to chromatin but not to DNA, and (b) the protein termed Su(var)3–7, which interacts with HP1 and chromatin (65). The principles underlying PEV in Drosophila seem also to apply to the stable inactivation of euchromatic genes needed for maintaining differential expression of developmental regulators (eg, the homeotic genes that control the segmental identity of the insect body). Locally silenced heritable chromatin structures are generated by the assembly of multiprotein complexes that contain members of the Polycomb group (Pc-G) of proteins, which are homologues of HP1 and contain a “chromodomain” (66). The Pc-G genes are responsible for maintaining long-range repression of homeotic genes over multiple enhancers in the later stages of Drosophila development. Like HP1, Pc-G proteins appear not to bind DNA. However, specific DNA sequences that recruit Polycomb group proteins and associated silencing factors [Polycomb response elements; (PREs)] have been identified and appear to be essential for stabilizing determined expression states, which are usually (but not invariably) repressive (see Ref. 66). Various models have been proposed for the mechanism of repression, including propagation of a repressive array of interacting proteins over chromosomal domains (the “spreading model” by analogy with the “spreading” effect of heterochromatin into euchromatin in PEV), an alternative model has been proposed in which clusters of Pc-G proteins interact cooperatively with each other to create tethered loop domains of chromatin which ensure repression (67). Mammalian homologues of HP1-like proteins have been found, as have interacting partners, and it seems likely that the same mechanism of silencing, by assembly into repressive heterochromatin, may be used to regulate some developmentally pivotal mammalian genes whose leaky expression would be disastrous for the cell.
Mammalian X-Chromosome Inactivation is an extreme case of heterochromatin formation over a whole chromosome. The precise molecular basis is not known, but there are some notable features of the chromatin composition: hypoacetylation of histone H4; the presence of an unusual H2A variant, macroH2A; heavy methylation of the DNA; and the presence of an unusual large untranslated RNA, termed Xist, which is expressed from the inactive X-chromosome, and which is of prime importance in an early event in X-inactivation (68). It would not be too surprising if X-inactivation turned out to have at least some features in common with telomeric and position effect variegation in yeast and Drosophila.
7. Replication-Linked Chromatin Assembly
During S-phase of the cell cycle in eukaryotes, it is not just the DNA that undergoes replication, but the chromatin as a whole. Chromatin assembly is tightly coupled to DNA replication and the passage of the replication fork. It involves assembly of nucleosomes on daughter strands, followed by conversion of an initially irregularly spaced array of nucleosomes to a regular array—a process known as maturation. There has been recent progress in understanding the participants in both of these processes. Assembly involves, in equal amounts, both old (parental) histones in preexisting nucleosomes, which are recycled, and newly synthesized histones, which are apparently randomly segregated on to the two nascent duplexes. The temporal order of deposition of newly synthesized histones on to DNA reflects the structural organization of the nucleosome; it begins with the deposition of H3 and H4 to form the central organizing tetrameric core, followed by H2A and H2B as two dimers, and finally addition of H1.
The newly synthesized H4 deposited at the replication fork is diacetylated, at lysines 5 and 12, in all species so far examined (see Histone Acetylation). Newly synthesized H3 is apparently also modified (acetylated or phosphorylated?), although perhaps not in all cell types. The role, if any, played by acetylation per se in the assembly process is not well understood. Acetylation is transient and is erased by deacetylases shortly after deposition of histones on to the DNA; one possibility might be that any tendency for premature formation of internucleosome contacts is avoided by the acetylation. The deposition of acetylated H3 and H4 involves the participation of chromatin assembly factor 1 (CAF-1), (69), which acts as a core histone molecular chaperone. It forms a stable complex with newly synthesized acetylated H3 and H4 and somehow targets them specifically to the replication fork (70); the histone chaperone NAP1 appears to handle H2A and H2B in Drosophila (71). Other proteins may also be involved in the assembly process. CAF-1 purified from human cells contains three subunits, indicated by their mass in kDa: p48, p60, and p150; homologues exist in yeast and Drosophila. The small subunit (p48) of human CAF-1 is homologous to the regulatory subunit (p46) of the human B-type histone acetyltransferase (responsible for deposition-related acetylation); both subunits bind to histone H4 and, significantly, at a site that is accessible only in the free histone and not in chromatin (as shown by the structure of the nucleosome core particle). Strikingly, similar subunits also occur in chromatin remodeling machines (eg, the p55 subunit of Drosophila NURF) as noted, as well as in histone deacetylases, leading to the suggestion that these subunits target the
various complexes to their histone substrates in a manner that is regulated by nucleosomal DNA (72) . They are members of the highly conserved family of proteins that contain “WD repeats,” multiple distinctive sequence motifs that appear to be involved in protein–protein interactions. Because the site of interaction of CAF-1 with H4 is accessible only in the free histone, CAF-1 would have to be displaced before nucleosome assembly could occur; the interactions appear to be disrupted by ACF (ATP-requiring chromatin assembly and remodeling factor; see text above). ACF was initially identified as a factor capable of regularly spacing an irregularly spaced nucleosome array (51) and would be well-suited to this role during the maturation step of chromatin assembly. It is also potentially able to participate in the remodeling of nucleosomes that accompanies transcriptional activation.
8. Chromatin Reconstitution
Reconstitution in vitro of chromatin from its component parts, like any complex biochemical structure, would allow questions to be asked about structure–function relationships. Reconstitution of the nucleosome core is straightforward. It is achieved by gradual dialysis of a 1:1 molar ratio of histone octamer and 146 bp DNA from 2M NaCl to low salt (eg, 10 mM NaCl). Nucleosome core particles for X-ray crystallography were made in this way. The histones can be presented either as the intact octamer or as a 1:1 molar ratio of H2A–H2B dimers and (H3)2(H4)2 tetramers.
Long “chromatin” reconstituted in vitro by mixing histones at high ionic strength (eg, 2M NaCl), and then dialyzing to low ionic strength, shows close packing of octamers, irrespective of the presence of H1, which has no effect because it is the last histone to bind in this procedure. In attempts to determine what cellular factors determine nucleosome spacing (nucleosome repeat length), several cell-free extracts, from Xenopus eggs and oocytes and from Drosophila embryos, have been developed that will assemble plasmid DNA, irrespective of sequence, into “physiologically” spaced chromatin in an ATP-dependent fashion, independent of replication. Nucleosome spacing (and hence linker length) is increased by H1. The earliest extracts were from Xenopus eggs, which contain a large maternal histone pool, and led to the discovery of two acidic histone chaperone proteins, nucleoplasmin and N1/N2, which facilitated chromatin assembly in this system.. The true role of these proteins may be storage of the large histone pool in the egg. Whether similar proteins exist in somatic cells is unclear, although nucleoplasmin-like proteins have been reported. The Drosophila embryo extracts are the same ones that led to the discovery of CHRAC (53) and ACF (51) (see text above: Chromatin remodeling) which themselves have nucleosome spacing activity. Spacing may also have an electrostatic component, involving neutralization of charges on linker DNA (73), and a connection between nucleosome spacing and formation of higher-order structure, which is also ionic strength dependent, has been suggested.
A well-defined in vitro system starting with pure components, which has recently been further explored (74), will also assemble “properly spaced” chromatin, in an H1-dependent, ATP-independent manner. Irregularly spaced octamers are first deposited on the DNA by dialysis from high to low NaCl concentration, and then a more regularly spaced array is generated by incubation at “physiological ionic strength” (150 mM NaCl) with H1(H5) in the presence of polyglutamate (which probably binds histones reversibly and allows equilibrium to be reached in the histone positions on the DNA). Interestingly, the system is sensitive to the DNA sequence, and regular nucleosome arrays were generated on only about half of the ~2-kbp cloned chicken genomic sequences tested; the sequences tested (by Southern blotting) behaved in essentially the same way in chicken liver nuclei. It is argued (74) that genomic chromatin is a mosaic of less well-ordered regions and regular regions that are about 10 nucleosomes long and contain a nucleosome positioning sequence (or sequences), which effectively acts as a boundary (18) against which linker-histone-dependent nucleosome alignment can occur. It is possible that egg, oocyte, and embryo extracts, which appear to be indifferent to DNA sequence as well as ATP-dependent, are designed to assemble chromatin through rapid cycles of cell division, and not to be responsive, for example, to genomic signals that may be necessary to specify arrays of nucleosomes compatible with gene expression and regulation in somatic cells.
References
1. K. van Holde (1988) Chromatin, Springer-Verlag, New York.
2. A. P. Wolffe (1995) Chromatin; Structure and Function, 2nd ed., Academic Press, New York.
3. S. C. R. Elgin (ed.) (1995) Chromatin Structure and Gene Expression [Frontiers in Molecular Biology series (B. D. Hames and D. M. Glover, eds.)], Oxford University Press, Oxford, U.K.
4. R. D. Kornberg (1977) Annu. Rev. Biochem. 46, 931–954.
5.K. Luger, A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond (1997) Nature 389, 251-260.
6. Y.-B. Zhou et al. (1999) Nature (in press. (
7. A. A. Travers (1987) Trends Biochem. Sci. 12, 108–112.
8.T. E. Shrader and D. M. Crothers (1989) Proc. Natl Acad. Sci. USA 86, 7418–7422.
9. H. R. Widlund et al. (1997) J. Mol. Biol. 267, 807–817.
10. P. T. Lowary and J. Widom (1998) J. Mol. Biol. 276, 19–42.
11. H. Cao, H. R. Widlund, T. Simonsson, and M. Kubista (1998) J. Mol. Biol. 281, 253–260.
12. J. Widom (1996) J. Mol. Biol. 259, 579–588.
13. S. Muyldermans and A. A. Travers (1994) J. Mol. Biol. 235, 855–870.
14. A. Travers and H. Drew (1999) Biopolymers (in press. (
15. J. J. Hayes, D. J. Clark, and A. P. Wolffe (1991) Proc. Natl. Acad. Sci. USA 88, 6829–6833.
16. C. Spangenberg et al. (1988) J. Mol. Biol. 278, 725–739.
17. S. Tanaka, M. Zatchej, and F. Thoma (1992) EMBO J. 11, 1187.
18. R. D. Kornberg and L. Stryer (1988) Nucleic Acids Res. 16, 6677–6690.
19. J. Svaren and W. Horz (1997) Trends Biochem. Sci. 22, 93–97.
20. T. Archer, M. G. Cordingley, R. G. Wolford, and G. L. Hager (1991) Mol. Cell. Biol. 11, 688–698.
21. G. Panetta et al. (1999) J. Mol. Biol. (in press. (
22. P. Bouvet, S. Dimitrov, and A. P. Wolffe (1994) Genes Dev. 8, 1147–1159.
23. G. Meersseman, S. Pennings, and E. M. Bradbury (1992) EMBO J. 11, 2951–2959.
24. S. Pennings, G. Meersseman, and E. M. Bradbury (1994) Proc. Natl. Acad. Sci USA 91, 10275–10279.
25. K. Ura, J. J. Hayes, and A. P. Wolffe (1995) EMBO J. 14, 3752–3765.
26. D. J. Clark and T. Kimura (1990) J. Mol. Biol. 211, 883–896.
27.G. Felsenfeld and J. D. McGhee (1986) Structure of the 30 nm fibre. Cell 44, 375–377.
28. J. Widom (1989) Annu. Rev. Biophys. Biophys. Chem. 18, 365–395.
29. K. van Holde and J. Zlatanova (1996) Prog. Nucleic Acid Res. Mol. Biol. 52, 217–259.
30. V. Ramakrishnan (1997) Crit. Rev. Eukaryot. Gene Expr. 7, 215–230.
31. J. Widom (1998) Annu. Rev. Biophys. Biomol. Struct. 27, 285–327.
32. F. Thoma, T. Koller, and A. Klug (1979) J. Cell Biol. 83, 403–427.
33.V. Graziano, S. E. Gerchman, D. K. Schneider, and V. Ramakrishnan (1994) Nature 368, 351–354.
34. C. L. F. Woodcock, L.-L. Frado, and J. B. Rattner (1984) J. Cell Biol. 99, 42–52.
35.C. L. Woodcock and R. Horowitz (1995) Trends Cell Biol. 5, 272–277.
36. J. D. McGhee, J. M. Nickol, G. Felsenfeld, and D. C. Rau (1983) Cell 33, 831–841.
37. J. O. Thomas, C. Rees, and P. J. G. Butler (1986). Eur. J. Biochem. 154, 343–348.
38. R. T. Kamakaka and J. O. Thomas (1990). EMBO J. 9, 3997–4006.
39. R. D. Kornberg and Y. Lorch (1992) Annu Rev. Cell Biol. 8, 563–589.
40. G. Felsenfeld (1992) Nature 335, 219–224.
41. T. R. Hebbes, A. W. Thorne, and C. Crane-Robinson (1988) EMBO J. 7, 1395–1402.
42. P. Geyer (1997) Curr. Opin. Genet. Dev. 7, 242–248.
43. S. Elgin (1988) J. Biol. Chem. 263, 19259–19262.
44. K. J. Polach and J. Widom (1995) J. Mol. Biol. 254, 130–149.
45. K. J. Polach and J. Widom (1996) J. Mol. Biol. 258, 800–812.
46. J. Boyes et al. (1998) J. Mol. Biol. 279, 529–544.
47. G. H. Thomas and S. C. R. Elgin (1988) EMBO J. 7, 2191–2201.
48. V. M. Studitsky, D. J. Clark, and G. Felsenfeld (1994) Cell 76, 371–382.
49.T. Tsukiyama and C. Wu (1997) Curr. Opin. Genet. Dev. 7, 182–191.
50. M. J. Pazin and J. T. Kadonaga (1997) Cell 88, 737–740.
51. Ito, J. K. Tyler, and J. T. Kadonaga (1997) Genes to Cells 2, 593–600.
52. B. Cairns (1998) Trends Biochem. Sci. 23, 20–25.
53.P. D. Varga-Weisz and P. B. Becker (1998) Curr. Opin. Cell Biol. 10, 346–353.
54. M. A. Martinez-Balbas, T. Tsukiyama, D. G. Dula, and C. Wu (1998) Proc. Natl. Acad. Sci. USA 95, 132–137.
55.R. T. Utley, J. Cote, T. Owen-Hughes, and J. L. Workman (1997) J Biol. Chem. 272, 12642–12649.
56. J. Coté, C. L. Peterson, and J. L. Workman (1998) Proc. Natl. Acad. Sci USA 95, 4947–4952.
57. G. Schnitzler, S. Sif, and R. E. Kingston (1998) Cell 94 17–27.
58. Y. Lorch, B. R. Cairns, M. Zhang, and R. D. Kornberg (1998) Cell 94, 29–34.
59. G. Orphanides et al. (1998) Cell 92, 105–116.
60. X. Nan et al. (1998) Nature 393, 386–389.
61. P. L. Jones et al. (1998) Nat. Genet. 19, 187–191.
62. M. A. McArthur and J. O. Thomas (1996) EMBO J. 15, 1705–1714.
63. M. Grunstein (1998) Cell 93, 325–328.
64. M. Grunstein (1997) Nature 389, 349–352.
65. L. L. Wallrath (1998) Curr. Opin. Genet. Dev. 8, 147–153.
66. G. Cavalli and R. Paro (1998) Curr. Opin. Cell Biol. 10, 354–360.
67.V. Pirrotta (1998) Cell 93, 333–336.
68. A. M. Keohane, J. S. Lavender, L. P. O''Neill, and B. M. Turner (1998) Dev. Genet. 22, 65–73.
69. S. Smith and B. Stillman (1989) Cell 58 15–25.
70. A. Verreault, P. D. Kaufman, R. Kobayashim, and B. Stillman (1996) Cell 87, 95–104.
71.T. Ito, M. Bulger, R. Kobayashi, and J. T. Kadonaga (1996) Mol. Cell. Biol. 16, 3112–3124.
72. A. Verreault, P. D. Kaufman, R. Kobayashi, and B. Stillman (1997) Curr. Biol. 8, 96–108.
73. T. A. Blank and P. B. Becker (1995) J. Mol. Biol. 252, 305–313.
74. K. Liu and A. Stein (1997) J. Mol. Biol. 270, 559–573.
75. P. D. Gregory and W. Horz (1998) Eur. J. Biochem. 251, 9–18.

|
|
|
|
دخلت غرفة فنسيت ماذا تريد من داخلها.. خبير يفسر الحالة
|
|
|
|
|
|
|
ثورة طبية.. ابتكار أصغر جهاز لتنظيم ضربات القلب في العالم
|
|
|
|
|
|
|
بالصور: تزامنا مع ختام فعالياته.. ممثل المرجعية العليا يشارك في المحفل القرآني المركزي في الصحن الحسيني الشريف
|
|
|