آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Mucopolysaccharidoses |


|
|
Read More
Date: 1-10-2021
Date: 26-8-2021
Date: 2-9-2021
|
Mucopolysaccharidoses
The mucopolysaccharidoses are hereditary diseases (approximately 1:25,000 live births) caused by a deficiency of any one of the lysosomal hydrolases normally involved in the degradation of heparan sulfate and/or dermatan sulfate (Fig. 1). They are progressive disorders characterized by lysosomal accumulation of GAG in various tissues, causing a range of symptoms, such as skeletal and ECM deformities, and intellectual disability. All are autosomal-recessive disorders except Hunter syndrome, which is X linked. Children who are homozygous for any one of these diseases are apparently normal at birth and then gradually deteriorate. In severe deficiencies, death occurs in childhood.
There currently is no cure. Incomplete lysosomal degradation of GAG results in the presence of oligosaccharides in the urine. These fragments can be used to diagnose the specific mucopolysaccharidosis by identifying the structure present on the nonreducing end of the oligosaccharide, because that residue would have been the substrate for the missing enzyme. Diagnosis is confirmed by measuring the patient’s cellular level of the lysosomal hydrolases. Bone marrow and cord blood transplants, in which transplanted macrophages produce the enzymes that degrade GAG, have been used to treat Hurler and Hunter syndromes, with limited success. Enzyme replacement therapy is available for both syndromes but does not prevent neurologic damage.
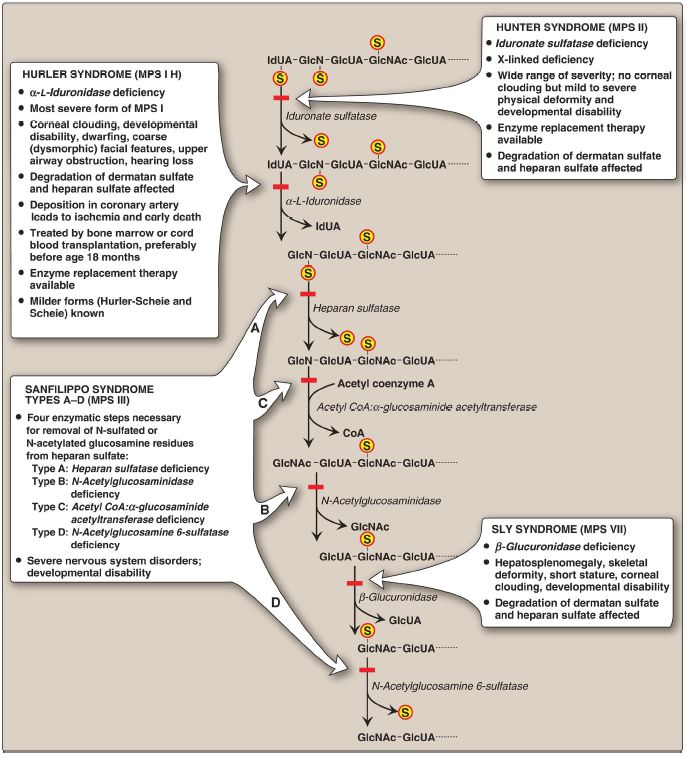
Figure 1: Degradation of the glycosaminoglycan heparan sulfate by lysosomal enzymes, indicating sites of enzyme deficiencies in some representative mucopolysaccharidoses (MPS). [Note: Deficiencies in galactosamine 6-sulfatase and β-galactosidase that degrade keratan sulfate result in Morquio syndrome (MPS IV), A and B, respectively. Deficiencies in arylsulfatase B that degrades dermatan sulfate result in Maroteaux-Lamy syndrome (MPS VI).] GlcUA and IdUA = glucuronic and iduronic acids; GalNAc = N-acetylgalactosamine; GlcNAc = N-acetylglucosamine; GlcN = glucosamine; S = sulfate.

|
|
|
|
علامات بسيطة في جسدك قد تنذر بمرض "قاتل"
|
|
|
|
|
|
|
أول صور ثلاثية الأبعاد للغدة الزعترية البشرية
|
|
|
|
|
|
|
مكتبة أمّ البنين النسويّة تصدر العدد 212 من مجلّة رياض الزهراء (عليها السلام)
|
|
|