























النبات

مواضيع عامة في علم النبات
الجذور - السيقان - الأوراق
النباتات الوعائية واللاوعائية
البذور (مغطاة البذور - عاريات البذور)
الطحالب
النباتات الطبية
الحيوان
مواضيع عامة في علم الحيوان
علم التشريح
التنوع الإحيائي
البايلوجيا الخلوية
الأحياء المجهرية
البكتيريا
الفطريات
الطفيليات
الفايروسات
علم الأمراض
الاورام
الامراض الوراثية
الامراض المناعية
الامراض المدارية
اضطرابات الدورة الدموية
مواضيع عامة في علم الامراض
الحشرات
التقانة الإحيائية
مواضيع عامة في التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحيوية والميكروبات
الفعاليات الحيوية
وراثة الاحياء المجهرية
تصنيف الاحياء المجهرية
الاحياء المجهرية في الطبيعة
أيض الاجهاد
التقنية الحيوية والبيئة
التقنية الحيوية والطب
التقنية الحيوية والزراعة
التقنية الحيوية والصناعة
التقنية الحيوية والطاقة
البحار والطحالب الصغيرة
عزل البروتين
هندسة الجينات
التقنية الحياتية النانوية
مفاهيم التقنية الحيوية النانوية
التراكيب النانوية والمجاهر المستخدمة في رؤيتها
تصنيع وتخليق المواد النانوية
تطبيقات التقنية النانوية والحيوية النانوية
الرقائق والمتحسسات الحيوية
المصفوفات المجهرية وحاسوب الدنا
اللقاحات
البيئة والتلوث
علم الأجنة
اعضاء التكاثر وتشكل الاعراس
الاخصاب
التشطر
العصيبة وتشكل الجسيدات
تشكل اللواحق الجنينية
تكون المعيدة وظهور الطبقات الجنينية
مقدمة لعلم الاجنة
الأحياء الجزيئي
مواضيع عامة في الاحياء الجزيئي
علم وظائف الأعضاء
الغدد
مواضيع عامة في الغدد
الغدد الصم و هرموناتها
الجسم تحت السريري
الغدة النخامية
الغدة الكظرية
الغدة التناسلية
الغدة الدرقية والجار الدرقية
الغدة البنكرياسية
الغدة الصنوبرية
مواضيع عامة في علم وظائف الاعضاء
الخلية الحيوانية
الجهاز العصبي
أعضاء الحس
الجهاز العضلي
السوائل الجسمية
الجهاز الدوري والليمف
الجهاز التنفسي
الجهاز الهضمي
الجهاز البولي
المضادات الميكروبية
مواضيع عامة في المضادات الميكروبية
مضادات البكتيريا
مضادات الفطريات
مضادات الطفيليات
مضادات الفايروسات
علم الخلية
الوراثة
الأحياء العامة
المناعة
التحليلات المرضية
الكيمياء الحيوية
مواضيع متنوعة أخرى
الانزيمات

Glycosaminoglycans, Proteoglycans, and Glycoproteins
 المؤلف:
Denise R. Ferrier
المؤلف:
Denise R. Ferrier
 المصدر:
Lippincott Illustrated Reviews: Biochemistry
المصدر:
Lippincott Illustrated Reviews: Biochemistry
 الجزء والصفحة:
الجزء والصفحة:
 3-10-2021
3-10-2021
 2431
2431




+
-
20
Glycosaminoglycans, Proteoglycans, and Glycoproteins
Glycosaminoglycans (GAG) are long, negatively charged, unbranched, heteropolysaccharide chains generally composed of a repeating disaccharide unit [acidic sugar–amino sugar]n (Fig. 1).
The amino sugar is either D-glucosamine or D-galactosamine in which the amino group is usually acetylated, thus eliminating its positive charge. The amino sugar may also be sulfated on carbon 4 or 6 or on a nonacetylated nitrogen. The acidic sugar is either D-glucuronic acid or its C-5 epimer L-iduronic acid.
GAG bind large amounts of water, thereby producing the gel-like matrix that forms the basis of the body’s ground substance. The viscous, lubricating properties of mucous secretions are also caused by the presence of GAG, which led to the original naming of these compounds as mucopolysaccharides. There are six major types of GAG, including chondroitin 4- and 6-sulfates, keratan sulfate, dermatan sulfate, heparin, heparan sulfate, and hyaluronic acid. All GAG, except hyaluronic acid, are found covalently attached to a core protein, forming proteoglycan monomers. Many proteoglycan monomers associate with a molecule of hyaluronic acid to form proteoglycan aggregates. GAG are synthesized in the Golgi. The polysaccharide chains are elongated by the sequential addition of alternating acidic and amino sugars, donated by their UDP derivatives. D-Glucuronate may be epimerized to L-iduronate. The last step in synthesis is sulfation of some of the amino sugars. The source of the sulfate is 3′-phosphoadenosyl-5′-phosphosulfate (PAPS). The completed proteoglycans are secreted into the extracellular matrix (ECM) or remain associated with the outer surface of cells.
GAG are degraded by lysosomal acid hydrolases. They are first broken down to oligosaccharides, which are degraded sequentially from the nonreducing end of each chain. A deficiency of any one of the hydrolases results in a mucopolysaccharidosis. These are hereditary disorders in which GAG accumulate in tissues, causing symptoms such as skeletal and ECM deformities and intellectual disability. Examples of these genetic diseases include Hunter (X-linked) and Hurler syndromes. Glycoproteins are proteins to which oligosaccharides (glycans) are covalently attached. They differ from the proteoglycans in that the length of the glycoprotein’s carbohydrate chain is relatively short (usually two to ten sugar residues long, although it can be longer), may be branched, and does not contain serial disaccharide units.
Membrane-bound glycoproteins participate in a broad range of cellular phenomena, including cell-surface recognition (by other cells, hormones, and viruses), cell-surface antigenicity (such as the blood group antigens), and as components of the ECM and of the mucins of the gastrointestinal and urogenital tracts, where they act as protective biologic lubricants. In addition, almost all of the globular proteins present in human plasma are glycoproteins. Glycoproteins are synthesized in the rough endoplasmic reticulum (RER) and the Golgi. The precursors of the carbohydrate components of glycoproteins are nucleotide sugars. O-Linked glycoproteins are synthesized in the Golgi by the sequential transfer of sugars from their nucleotide carriers to the hydroxyl group of a serine or threonine residue in the protein.
N-Linked glycoproteins are synthesized by the transfer of a preformed oligosaccharide from its RER membrane lipid carrier, dolichol pyrophosphate, to the amide N of an asparagine residue in the protein. They contain varying amounts of mannose. A deficiency in the phosphotransferase that phosphorylates mannose residues at carbon 6 in N-linked glycoprotein enzymes destined for the lysosomes results in I-cell disease. Glycoproteins are degraded in lysosomes by acid hydrolases.
A deficiency of any one of these enzymes results in a lysosomal glycoprotein storage disease (oligosaccharidosis), resulting in accumulation of partially degraded structures in the lysosome.
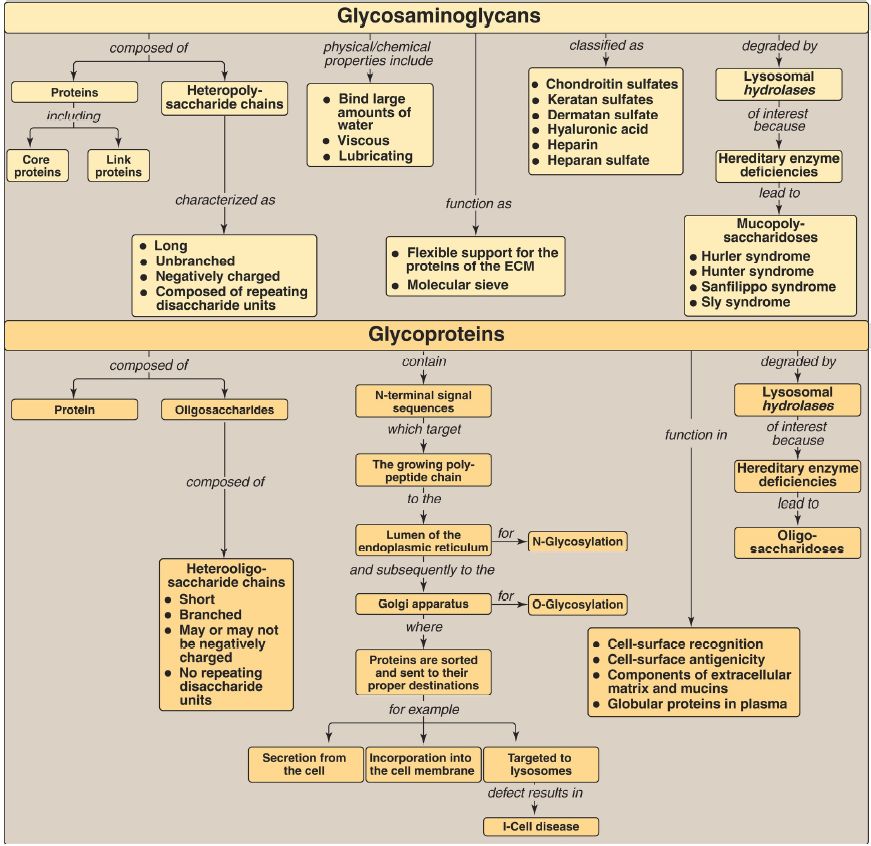
Figure 1: Key concept map for glycosaminoglycans and glycoproteins. ECM = extracellular matrix.
 الاكثر قراءة في الكيمياء الحيوية
الاكثر قراءة في الكيمياء الحيوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















