























النبات

مواضيع عامة في علم النبات
الجذور - السيقان - الأوراق
النباتات الوعائية واللاوعائية
البذور (مغطاة البذور - عاريات البذور)
الطحالب
النباتات الطبية
الحيوان
مواضيع عامة في علم الحيوان
علم التشريح
التنوع الإحيائي
البايلوجيا الخلوية
الأحياء المجهرية
البكتيريا
الفطريات
الطفيليات
الفايروسات
علم الأمراض
الاورام
الامراض الوراثية
الامراض المناعية
الامراض المدارية
اضطرابات الدورة الدموية
مواضيع عامة في علم الامراض
الحشرات
التقانة الإحيائية
مواضيع عامة في التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحيوية والميكروبات
الفعاليات الحيوية
وراثة الاحياء المجهرية
تصنيف الاحياء المجهرية
الاحياء المجهرية في الطبيعة
أيض الاجهاد
التقنية الحيوية والبيئة
التقنية الحيوية والطب
التقنية الحيوية والزراعة
التقنية الحيوية والصناعة
التقنية الحيوية والطاقة
البحار والطحالب الصغيرة
عزل البروتين
هندسة الجينات
التقنية الحياتية النانوية
مفاهيم التقنية الحيوية النانوية
التراكيب النانوية والمجاهر المستخدمة في رؤيتها
تصنيع وتخليق المواد النانوية
تطبيقات التقنية النانوية والحيوية النانوية
الرقائق والمتحسسات الحيوية
المصفوفات المجهرية وحاسوب الدنا
اللقاحات
البيئة والتلوث
علم الأجنة
اعضاء التكاثر وتشكل الاعراس
الاخصاب
التشطر
العصيبة وتشكل الجسيدات
تشكل اللواحق الجنينية
تكون المعيدة وظهور الطبقات الجنينية
مقدمة لعلم الاجنة
الأحياء الجزيئي
مواضيع عامة في الاحياء الجزيئي
علم وظائف الأعضاء
الغدد
مواضيع عامة في الغدد
الغدد الصم و هرموناتها
الجسم تحت السريري
الغدة النخامية
الغدة الكظرية
الغدة التناسلية
الغدة الدرقية والجار الدرقية
الغدة البنكرياسية
الغدة الصنوبرية
مواضيع عامة في علم وظائف الاعضاء
الخلية الحيوانية
الجهاز العصبي
أعضاء الحس
الجهاز العضلي
السوائل الجسمية
الجهاز الدوري والليمف
الجهاز التنفسي
الجهاز الهضمي
الجهاز البولي
المضادات الميكروبية
مواضيع عامة في المضادات الميكروبية
مضادات البكتيريا
مضادات الفطريات
مضادات الطفيليات
مضادات الفايروسات
علم الخلية
الوراثة
الأحياء العامة
المناعة
التحليلات المرضية
الكيمياء الحيوية
مواضيع متنوعة أخرى
الانزيمات

Amino Acid Metabolism
 المؤلف:
Denise R. Ferrier
المؤلف:
Denise R. Ferrier
 المصدر:
Lippincott Illustrated Reviews: Biochemistry
المصدر:
Lippincott Illustrated Reviews: Biochemistry
 الجزء والصفحة:
الجزء والصفحة:
 12-11-2021
12-11-2021
 2246
2246




+
-
20
Amino Acid Metabolism
Amino acids whose catabolism yields pyruvate or an intermediate of the tricarboxylic acid cycle are termed glucogenic (Fig. 1). They can give rise to the net formation of glucose in the liver and kidneys. The solely glucogenic amino acids are glutamine, glutamate, proline, arginine, histidine, alanine, serine, glycine, cysteine, methionine, valine, threonine, aspartate, and asparagine.
Amino acids whose catabolism yields either acetoacetate or one of its precursors, acetyl coenzyme A (CoA) or acetoacetyl CoA, are termed ketogenic. Leucine and lysine are solely ketogenic. Tyrosine, phenylalanine, tryptophan, and isoleucine are both ketogenic and glucogenic. Nonessential amino acids can be synthesized from metabolic intermediates or from the carbon skeletons of essential amino acids. Essential amino acids need to be obtained from the diet. They include histidine, methionine, threonine, valine, isoleucine, phenylalanine, tryptophan, leucine, and lysine. Phenylketonuria (PKU) is caused by a deficiency of phenylalanine hydroxylase (PAH), which converts phenylalanine to tyrosine. Hyperphenylalaninemia may also be caused by deficiencies in the enzymes that synthesize or regenerate the coenzyme for PAH, tetrahydrobiopterin. Untreated individuals with PKU suffer from severe intellectual disability, developmental delay, microcephaly, seizures, and a characteristic musty (mousy) smell of the urine.
Treatment involves controlling dietary phenylalanine. Tyrosine becomes an essential dietary component for people with PKU. Maple syrup urine disease (MSUD) is caused by a partial or complete deficiency in branched-chain a-keto acid dehydrogenase, the enzyme that decarboxylates the branched-chain amino acids (BCAA) leucine, isoleucine, and valine. Symptoms include feeding problems, vomiting, ketoacidosis, changes in muscle tone, and a characteristic sweet smell of the urine. If untreated, the disease leads to neurologic problems that result in death. Treatment involves controlling BCAA intake. Other important genetic diseases associated with amino acid metabolism include albinism, homocystinuria, methylmalonic acidemia, alkaptonuria, histidinemia, tyrosinemia, and cystathioninuria.
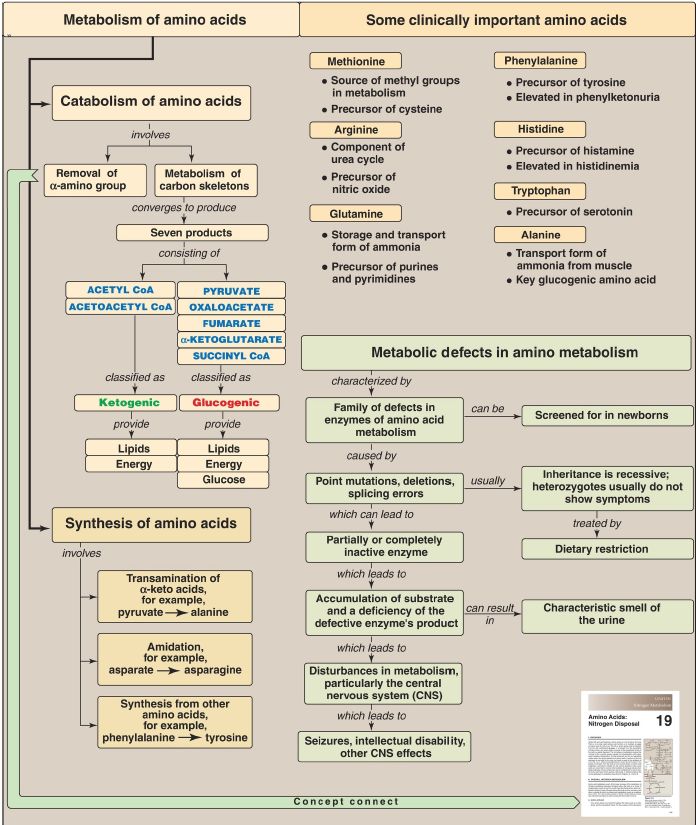
Figure 1: Key concept map for amino acid metabolism. CoA = coenzyme A.
 الاكثر قراءة في الكيمياء الحيوية
الاكثر قراءة في الكيمياء الحيوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















