























النبات

مواضيع عامة في علم النبات
الجذور - السيقان - الأوراق
النباتات الوعائية واللاوعائية
البذور (مغطاة البذور - عاريات البذور)
الطحالب
النباتات الطبية
الحيوان
مواضيع عامة في علم الحيوان
علم التشريح
التنوع الإحيائي
البايلوجيا الخلوية
الأحياء المجهرية
البكتيريا
الفطريات
الطفيليات
الفايروسات
علم الأمراض
الاورام
الامراض الوراثية
الامراض المناعية
الامراض المدارية
اضطرابات الدورة الدموية
مواضيع عامة في علم الامراض
الحشرات
التقانة الإحيائية
مواضيع عامة في التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحيوية والميكروبات
الفعاليات الحيوية
وراثة الاحياء المجهرية
تصنيف الاحياء المجهرية
الاحياء المجهرية في الطبيعة
أيض الاجهاد
التقنية الحيوية والبيئة
التقنية الحيوية والطب
التقنية الحيوية والزراعة
التقنية الحيوية والصناعة
التقنية الحيوية والطاقة
البحار والطحالب الصغيرة
عزل البروتين
هندسة الجينات
التقنية الحياتية النانوية
مفاهيم التقنية الحيوية النانوية
التراكيب النانوية والمجاهر المستخدمة في رؤيتها
تصنيع وتخليق المواد النانوية
تطبيقات التقنية النانوية والحيوية النانوية
الرقائق والمتحسسات الحيوية
المصفوفات المجهرية وحاسوب الدنا
اللقاحات
البيئة والتلوث
علم الأجنة
اعضاء التكاثر وتشكل الاعراس
الاخصاب
التشطر
العصيبة وتشكل الجسيدات
تشكل اللواحق الجنينية
تكون المعيدة وظهور الطبقات الجنينية
مقدمة لعلم الاجنة
الأحياء الجزيئي
مواضيع عامة في الاحياء الجزيئي
علم وظائف الأعضاء
الغدد
مواضيع عامة في الغدد
الغدد الصم و هرموناتها
الجسم تحت السريري
الغدة النخامية
الغدة الكظرية
الغدة التناسلية
الغدة الدرقية والجار الدرقية
الغدة البنكرياسية
الغدة الصنوبرية
مواضيع عامة في علم وظائف الاعضاء
الخلية الحيوانية
الجهاز العصبي
أعضاء الحس
الجهاز العضلي
السوائل الجسمية
الجهاز الدوري والليمف
الجهاز التنفسي
الجهاز الهضمي
الجهاز البولي
المضادات الميكروبية
مواضيع عامة في المضادات الميكروبية
مضادات البكتيريا
مضادات الفطريات
مضادات الطفيليات
مضادات الفايروسات
علم الخلية
الوراثة
الأحياء العامة
المناعة
التحليلات المرضية
الكيمياء الحيوية
مواضيع متنوعة أخرى
الانزيمات

Blotting Methods
 المؤلف:
JOCELYN E. KREBS, ELLIOTT S. GOLDSTEIN and STEPHEN T. KILPATRICK
المؤلف:
JOCELYN E. KREBS, ELLIOTT S. GOLDSTEIN and STEPHEN T. KILPATRICK
 المصدر:
LEWIN’S GENES XII
المصدر:
LEWIN’S GENES XII
 الجزء والصفحة:
الجزء والصفحة:
 7-3-2021
7-3-2021
 4453
4453




+
-
20
Blotting Methods
KEY CONCEPTS
- Southern blotting involves the transfer of DNA from a gel to a membrane, followed by detection of specific sequences by hybridization with a labeled probe.
- Northern blotting is similar to Southern blotting but involves the transfer of RNA from a gel to a membrane.
- Western blotting entails separation of proteins on a sodium dodecyl sulfate (SDS) gel, transfer to a nitrocellulose membrane, and detection of proteins of interest using antibodies.
After nucleic acids are separated by size in a gel matrix, they can be detected using dyes that are sequence-nonspecific, or specific sequences can be detected using a method generically referred to as blotting. Although slower and more involved than direct visualization by fluorescent dye staining, blotting techniques have two major advantages: They have a greatly increased sensitivity relative to dye staining, and they allow for the specific detection of defined sequences of interest among many similarly sized bands on a gel.
The method was first developed for application to DNA agarose gels and was briefly introduced in the section Nucleic Acid Detection. In this form, the method is referred to as Southern blotting (after the method’s inventor, Dr. Edwin Southern). A schematic of this process is shown in FIGURE 1. A regular agarose gel is made and run (and if desired, stained) as described previously. Following this, the gel is soaked in an alkali buffer to denature the DNA, and then placed in contact with a sheet of porous membrane (commonly nitrocellulose or nylon). Next, a buffer is drawn through the gel and then the membrane either by capillary action (e.g., by wicking into a stack of dry paper towel) or by a gentle vacuum pressure. This slow flow of buffer in turn draws each nucleic acid band in the gel out of the gel matrix and onto the membrane surface. Nucleic acids bind to the membrane, which in many cases is positively charged to increase efficiency of DNA binding. This, in effect, creates a “contact print” of the order and position of all nucleic acid bands as size-resolved in the gel. To make the elution of large DNA molecules from the gel matrix more efficient, the gel is sometimes treated with a mild acid after electrophoresis but before transfer. This induces nucleic acid depurination and creates random strand breaks in the DNA within the gel, such that large molecules are broken into smaller subsections that elute more readily but remain in the same physical location as their original gel band.

FIGURE 1. To perform a Southern blot, DNA digested with restriction enzymes is electrophoresed to separate fragments by size. Double-stranded DNA is denatured in an alkali solution either before or during blotting. The gel is placed on a wick (such as a sponge) in a container of transfer buffer and a membrane (nylon or nitrocellulose) is placed on top of the gel. Absorbent materials such as paper towels are placed on top. Buffer is drawn from the reservoir through the gel by capillary action, transferring the DNA to the membrane. The membrane is then incubated with a labeled probe (usually DNA). The unbound probe is washed away, and the bound probe is detected by autoradiography or phosphorimaging. In Northern blotting, RNA is run on a gel rather than DNA.
Following transfer, the nucleic acids are fixed to the membrane either through drying or through exposure to ultraviolet light, which can create physical crosslinks between the membrane and the nucleic acids (primarily pyrimidines). The blot is now ready for blocking, where it is immersed in a warmed, low-salt buffer containing materials that will bind to and block areas of the blot that might bind organic compounds nonspecifically. Following blocking, a probe molecule is introduced. The probe consists of a labeled (isotopically or chemically, e.g., through incorporation of biotinylated nucleotides) copy of the target sequence of interest, which is either synthesized as a single-stranded oligonucleotide, or (if double stranded) has been heat denatured and rapidly cooled to place it in a single-stranded form. When this is added to the warmed buffer and allowed to incubate with the blocked membrane, the probe will attempt to hybridize to homologous sequences on the membrane surface. Following this hybridization step, the membrane is generally washed in warm buffer without a
probe or blocking agent to remove nonspecifically associated probe molecules, and then visualized; in the case of isotopically labeled probes, this can be done by simply exposing the membrane to a piece of film or a phosphor-imager screen. Decay of the label (usually 32P or 35S) leads to the production of an image in which any hybridized DNA bands become visible on the developed film or scanned phosphor screen. For chemically labeled probes, chemiluminescent or fluorescent detection strategies are used in an analogous manner.
A final benefit of the Southern blotting technique is that the observed band intensity is related to the amount of target on the membrane—in other words, it is a quantitative method. If a suitable standard (e.g., a dilution series of unlabeled probe sequence) is included in the gel, comparison of this standard to target band intensities allows for determination of target quantity in the starting sample. This information can be useful for applications such as determining viral copy number in a host cell sample.
Numerous variations on the Southern-blot approach exist, including use of specialized gel systems for the initial separation of DNAs. For example, two-dimensional gels can be used to separate DNA molecules by shape as well as size. FIGURE 2. illustrates a twodimensional mapping technique used to identify replication intermediates, a method used extensively in studies of replication and replication repair. In this method, restriction fragments of replicating DNA are electrophoresed in a first dimension that separates by mass and a second dimension where movement is determined more by shape. Different types of replicating molecules follow characteristic paths, measured by their deviation from the line that would be followed by a linear molecule of DNA that doubled in size. A simple Y-structure (which occurs when a fragment is in the midst of replication, but does not itself contain an origin of replication) follows a continuous path in which one fork moves along the linear fragment. An inflection point occurs when all three branches are the same length and the structure therefore deviates most extensively from linear DNA. Analogous considerations determine the paths of double Y-structures or bubbles (bubbles indicate a bidirectional fork, thus an origin of replication, within the fragment). An asymmetric bubble follows a discontinuous path, with a break at the point at which the bubble is converted to a Y-structure as one fork runs off the end.
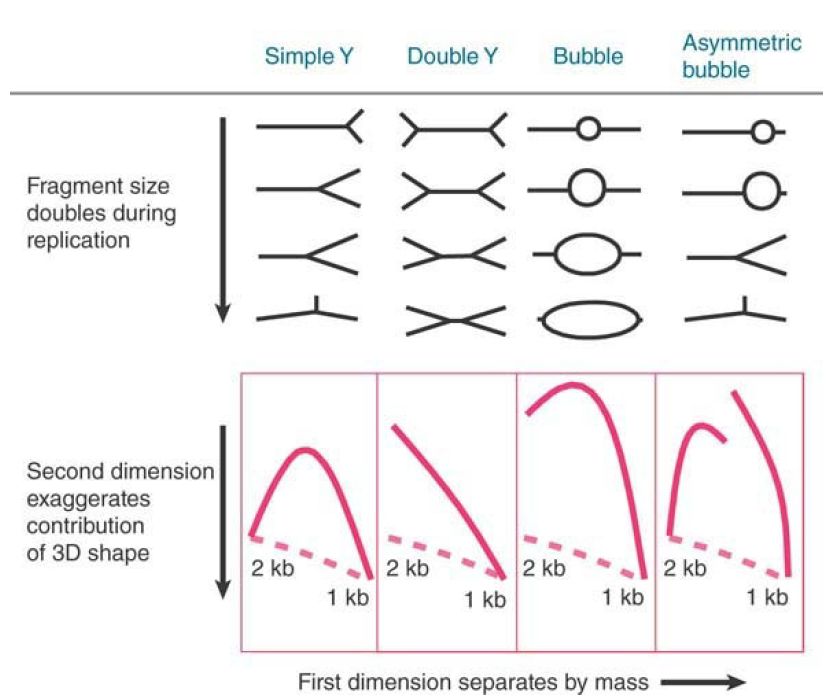
FIGURE 2. One application of Southern blotting allows detection of fragments separated by shape as well as size. In this example, the position of a replication origin and the number of replicating forks determine the shape of a replicating restriction fragment, which can be followed by its electrophoretic path (solid line). The dashed line shows the path for a linear DNA.
Another variation of the Southern-blot approach is the use of a denaturing gel matrix for an otherwise analogous process on RNA molecules (referred to as northern blotting). In this case, there is no initial digestion step, so intact RNA molecules are separated by size, usually on a formaldehyde or other denaturing gel, which eliminates RNA secondary structures. This allows measurement of actual RNA sizes and, like Southern blotting, provides a similarly quantitative method for detection of any type of RNA. If mRNA is the target of interest, it is possible to separate mRNA from all the other classes of RNA in the cell. mRNA (and some noncoding RNA) differs from other RNAs in that it is polyadenylated . Poly(A)+ mRNA can therefore be enriched by use of an oligo(dT) column, in which oligomers of oligo(dT) are immobilized on a solid support and used to capture mRNA from the total RNA in a sample. This is illustrated in FIGURE2.
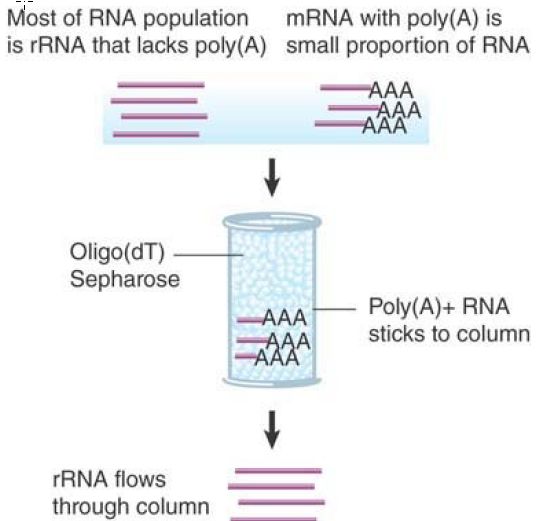
FIGURE 3. Poly(A)+ RNA can be separated from other RNAs by fractionation on an oligo(dT) column.
A conceptually similar process for proteins based on proteinseparation gels and blotting to membrane is known as western blotting. This method is depicted in FIGURE 4. There are some key differences between the procedures for blotting proteins compared to nucleic acids. First, protein-separation gels typically contain the detergent SDS, which serves to unfold the proteins so that they will migrate according to size rather than shape. It also provides a uniform negative charge to all proteins so that they will migrate toward the positive pole of the gel. (In the absence of SDS, each protein has a specific individual charge at a given pH; it is possible to separate proteins based on these charges, rather than size, in a technique called isoelectric focusing.)
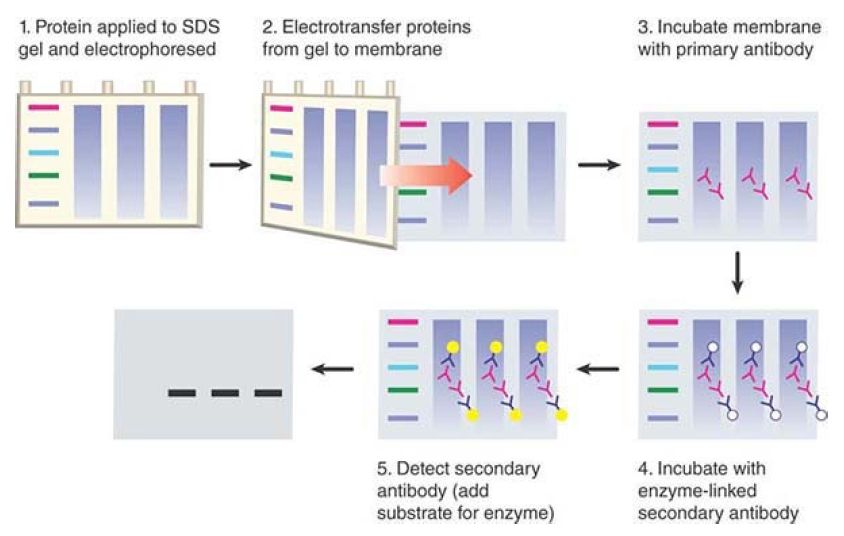
FIGURE 4. In a western blot, proteins are separated by size on an SDS gel, transferred to a nitrocellulose membrane, and detected by using an antibody. The primary antibody detects the protein and the enzyme-linked secondary antibody detects the primary antibody. The secondary antibody is detected in this example via addition of a chemiluminescent substrate, which results in emission of light that can be detected on X-ray film.
After the proteins are separated on the gel, they are transferred to a nitrocellulose membrane using an electric current to effect the transfer, rather than the capillary or vacuum methods used for nucleic acids. The most significant difference in western blotting is the method of detecting proteins on the membrane.
Complementary base pairing can’t be used to detect a protein, so westerns use antibodies to recognize the protein of interest. The antibody can either recognize the protein itself, if such an antibody is available, or it can recognize an epitope tag that has been fused to the protein sequence. An epitope tag is a short peptide sequence that is recognized by a commercially available antibody; the DNA encoding the tag can be cloned in-frame to a gene of interest, resulting in a product containing the epitope (typically at the N- or C-terminus of the protein). Sequences for the most commonly used epitope tags (such as the HA, FLAG, and myc tags) are often available in expression vectors for ease of fusion .
The antibody that recognizes the target on the membrane is known as the primary antibody. The final stage of western blotting is detection of the primary antibody with a secondary antibody, which is the antibody that can be visualized. Secondary antibodies are raised in a different species from the primary antibody used and recognize the constant region of the primary antibody (e.g., a “goat antirabbit” antibody will recognize a primary antibody raised in a rabbit; see the chapter titled Somatic DNA Recombination and Hypermutation in the Immune System for a review of antibody structure). The secondary antibody is typically linked to a moiety that allows its visualization—for example, a fluorescent dye or an enzyme such as alkaline phosphatase or horseradish peroxidase.These enzymes serve as visualization tools because they can convert added substrates to a colored product (colorimetric detection) or can release light as a reaction product (chemiluminescent detection). Use of primary and secondary
antibodies (rather than linking a visualizer to the primary antibody) increases the sensitivity of western blotting. The result is semiquantitative detection of the protein of interest.
Continuing in the same vein, techniques used to identify interactions between DNA and proteins (through protein gel separation and blotting followed by probing with a DNA) are southwestern blotting; when an RNA probe is used, the technique is northwestern blotting.
 الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















