























النبات

مواضيع عامة في علم النبات
الجذور - السيقان - الأوراق
النباتات الوعائية واللاوعائية
البذور (مغطاة البذور - عاريات البذور)
الطحالب
النباتات الطبية
الحيوان
مواضيع عامة في علم الحيوان
علم التشريح
التنوع الإحيائي
البايلوجيا الخلوية
الأحياء المجهرية
البكتيريا
الفطريات
الطفيليات
الفايروسات
علم الأمراض
الاورام
الامراض الوراثية
الامراض المناعية
الامراض المدارية
اضطرابات الدورة الدموية
مواضيع عامة في علم الامراض
الحشرات
التقانة الإحيائية
مواضيع عامة في التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحيوية والميكروبات
الفعاليات الحيوية
وراثة الاحياء المجهرية
تصنيف الاحياء المجهرية
الاحياء المجهرية في الطبيعة
أيض الاجهاد
التقنية الحيوية والبيئة
التقنية الحيوية والطب
التقنية الحيوية والزراعة
التقنية الحيوية والصناعة
التقنية الحيوية والطاقة
البحار والطحالب الصغيرة
عزل البروتين
هندسة الجينات
التقنية الحياتية النانوية
مفاهيم التقنية الحيوية النانوية
التراكيب النانوية والمجاهر المستخدمة في رؤيتها
تصنيع وتخليق المواد النانوية
تطبيقات التقنية النانوية والحيوية النانوية
الرقائق والمتحسسات الحيوية
المصفوفات المجهرية وحاسوب الدنا
اللقاحات
البيئة والتلوث
علم الأجنة
اعضاء التكاثر وتشكل الاعراس
الاخصاب
التشطر
العصيبة وتشكل الجسيدات
تشكل اللواحق الجنينية
تكون المعيدة وظهور الطبقات الجنينية
مقدمة لعلم الاجنة
الأحياء الجزيئي
مواضيع عامة في الاحياء الجزيئي
علم وظائف الأعضاء
الغدد
مواضيع عامة في الغدد
الغدد الصم و هرموناتها
الجسم تحت السريري
الغدة النخامية
الغدة الكظرية
الغدة التناسلية
الغدة الدرقية والجار الدرقية
الغدة البنكرياسية
الغدة الصنوبرية
مواضيع عامة في علم وظائف الاعضاء
الخلية الحيوانية
الجهاز العصبي
أعضاء الحس
الجهاز العضلي
السوائل الجسمية
الجهاز الدوري والليمف
الجهاز التنفسي
الجهاز الهضمي
الجهاز البولي
المضادات الميكروبية
مواضيع عامة في المضادات الميكروبية
مضادات البكتيريا
مضادات الفطريات
مضادات الطفيليات
مضادات الفايروسات
علم الخلية
الوراثة
الأحياء العامة
المناعة
التحليلات المرضية
الكيمياء الحيوية
مواضيع متنوعة أخرى
الانزيمات

Genome Projects Background
 المؤلف:
John M Walker and Ralph Rapley
المؤلف:
John M Walker and Ralph Rapley
 المصدر:
Molecular Biology and Biotechnology 5th Edition
المصدر:
Molecular Biology and Biotechnology 5th Edition
 الجزء والصفحة:
الجزء والصفحة:
 27-11-2020
27-11-2020
 2330
2330




+
-
20
Genome Projects Background
The Human Genome Project was conceived in the mid-1980s, but was officially launched in 1990. This led to an upsurge in DNA sequencing activity and an avalanche of sequence data in a variety of species. By 1995, the first cellular (as opposed to viral) genome of a bacterium had been completed and this was followed by a stream of sequenced genomes, from prokaryotes initially and then eukaryotes. Two independent drafts of the human genome euchromatic DNA sequence were published simultaneously in 2001 as a result of a race between by the publicly funded International Human Genome Project and the privately funded initiative led by Celera Genomics. These groups estimated that the human genome consists of between 30 500 and 35 5006 and between 26 000 and 38 0007 expressed genes. Today this is considered an overestimate, the actual number being closer to 20 000–25 000 genes, very much lower than the pre-genome project estimates of 80 000–140 000 genes. Rapid and powerful advances in cloning and sequencing technologies and computational biology have transformed genome sequencing initiatives from massive long-term endeavours to relatively quick and much less expensive undertakings. By mid-2008, complete genome sequences had been generated for 809 species, including 94 complex eukaryotic genomes.76 There are currently more than 2500 genomes under study and the number is set to grow as the precision and speed of genome sequencing technologies improve and the cost is reduced. It is estimated that the financial cost of the Human Genome Project, which involved a huge international effort over more than 10 years, was US $500 million, but it is predicted that the cost of resequencing any individual’s entire DNA sequence could soon be just $1000. This would bring human genomic analysis into the realms of personal medicine.
Mapping and Sequencing Strategies
The Human Genome Project aimed to produce four types of map: physical, genetic, DNA sequence and gene. Physical and genetic maps provide essential anchor points and frameworks to align DNA sequences and assign genes. A high-resolution physical map based on the analysis of overlapping DNA clones represents the actual distance in DNA base pairs between genetic markers and other landmarks. However, the ultimate physical map is the DNA sequence itself. Low-resolution physical maps are generated from techniques such as somatic cell hybridisation and fluorescence in situ hybridisation.
There are two approaches to genome sequencing: whole genome shotgun sequencing (WGS) and the more labour-intensive hierarchical shotgun sequencing (HS). In simple organisms such as bacteria and viruses, where the chromosomes are haploid and very little repeat sequence occurs, or for sequencing individual human genes, WGS works well.77,78 In contrast, for eukaryotic genomes, where repeat sequences often abound, including the human genome (450% repeats), and there is considerable heterozygosity, it has been argued that HS offers advantages over WGS, and this was the approach adopted by the publicly funded International Human Genome Sequencing Consortium.
1.1 Hierarchical Sequencing
This approach relies on the production of a set of large-insert clones (typically 100–200 kb each) that cover the entire genome.Since the clones are all independent and ultimately positioned on a physical map in an order that represents each chromosome, repeated sequences are far less troublesome, leading to fewer gaps than encountered by the WGS approach. For many genome projects, high-capacity vectors such as BACs are advantageously used for generating large-insert clones since they are less likely to rearrange than alternatives such as YACs. Longrange physical maps are generated from the production of ‘contigs’.
Contigs are a set of overlapping DNA fragments that have been obtained from independent clones and positioned relative to one another so that they form a contiguous array. To obtain contigs, genomic libraries must prepared from high molecular weight DNA that has either been partially digested with restriction enzymes or randomly sheared. Partial digestion or random shearing leads to the production of a set of overlapping clones, whereas complete digestion would produce a set of fragments with no overlaps (Figure 1). Partial digestion ensures that when each DNA fragment is cloned into a vector, it has ends that will overlap with other clones. Thus, when the overlaps are identified, the clones can be positioned or ordered, so that a physical map is produced.
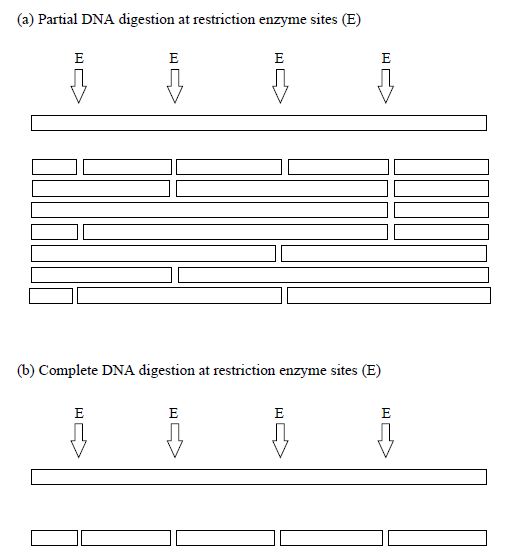
Figure 1 Comparison of partial and complete digestion of DNA molecules at restriction enzymes sites (E).
Large-insert clones are broken down further into sets of smaller overlapping subclones and sequenced using the shotgun sequencing approach. In order to position the overlapping ends into a contig representing the large insert clone, it is preferable to undertake DNA sequencing of both ends of the individual subclones (double-barrelled
shotgun sequencing). Eventually, the entire DNA sequence of the largeinsert clone is obtained by computer-based alignment of individual subclone DNA end sequences. In order to minimise the overlaps and identify the large-insert clones for further sequencing, restriction enzyme mapping can be undertaken to produce a ‘fingerprint clone contig’. In the Human Genome Project, fingerprint clone contigs were mapped to
human chromosomal locations by each chromosome workgroup using resources such as panels of human radiation hybrids (RH), fluorescence in situ hybridisation (FISH) with human chromosomes and existing genetic maps. Radiation hybrids are panels of human–hamster cell hybrids formed by fusing human cells containing radiation-generated fragments of human chromosomes with hamster cells. Panels of radiation hybrids that contain characterized fragments from all human chromosomes can be used for constructing genetic maps that are complementary to both recombination maps and physical maps based on contigs. In order to define a common way for all research laboratories to order clones and connect physical maps together, an arbitrary molecular technique based on the PCR has been developed to generate sequencetagged sites (STS). These are small, unique sequences between 200 and 300 base pairs that are amplified by PCR.80 The uniqueness of the STS is defined by the PCR primers that flank the STS. If the PCR results in amplification, then the STS is present in the clone being tested. In this way, defining STS markers that lie approximately 100 kb apart along a contig map allows the ordering of those contigs. Thus, all groups working with clones have publicly available defined landmarks with which to order clones produced in their DNA libraries (Figure 2).
STSs may also be generated from polymorphic markers that may be traced through families along with other DNA markers and located on a genetic linkage map. These polymorphic STSs may thus serve as markers on both a physical map and a genetic linkage map for each chromosome and therefore provide a useful means for aligning the two types of map.
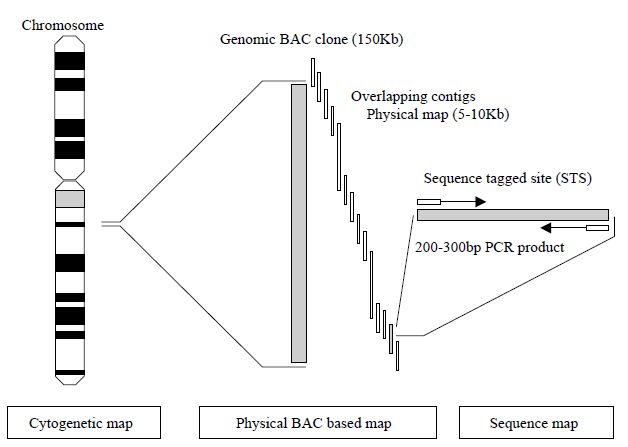
Figure 2. Schematic of the use of STS markers in the hierarchical physical mapping
a human chromosome using BAC clones.
In addition to the human genome, the hierarchical sequencing approach has been used to sequence several genomes, including those of the yeast Saccharomyces cerevisiae and the nematode worm Cae- norhabditis elegans.High-quality BAC clone-based physical genome maps in one species can be of great value to genome projects in other species where some conservation of genomic sequence and gene order might be expected. For example, the outputs of the Human Genome Project have provided anchors for ordering BAC clones generated in several other species such as mouse, rat and cattle, allowing simplification of clone alignments and physical map building in addition to the generation of comparative maps in the respective species. The International Human Genome Sequencing Consortium sequence was reported as finished in 2004 (Build 35) and contains 2.85 billion nucleotides interrupted by 341 gaps. It covers approximately 99% of the euchromatic genome.
1.2 Whole Genome Shotgun Sequencing (WGS)
In contrast to the hierarchical BAC by BAC approach, which relies on the availability of genetic and physical maps for success, WGS is based on the strategy of sequencing a vast number of random genomic clones followed by intensive computer-based analysis of the DNA sequences which identifies matching sequences in different clones. This permits the assembly of a chromosomal DNA sequence, in principle without other map resources. As with the HS approach, overlapping clones are required, but since the clones are destined for direct sequence analysis, only vectors that contain small to medium inserts are normally used. Hence once the overlaps have been identified, the entire sequence is assembled. WGS was the approach adopted by the privately funded human genome initiative.
Although WGS remains somewhat controversial for sequencing complex genomes of ‘higher’ organisms because of the problems associated with repeat sequences and heterozygosity, it is a widely used approach. The number of complex genomes sequenced by this WGS is increasing and includes the fruit fly Drosophila, mosquito (anopheles), mouse, puffer fish, dog and grapevine. However, in some cases, such as the silk worm genome project, the WGS method has resulted in many seemingly irresolvable gaps in the genome and so the BAC-based hierarchical ordering of clones was used to close the gaps.93Advances in computational analysis of WGS sequences suggest that the problems caused by repeat sequences could be overcome, hence the approach can be expected to gain more ground in future genome projects.
 الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















